基于规范化诊治与管理的234例儿童癫性脑病病例系列报告
2019-03-28李奕洁杜晓楠吴冰冰姜永辉陈光福
李奕洁 杜晓楠 吴冰冰 姜永辉 郑 静 陈光福,5 王 艺,5

1 方法
1.2 EE病例纳入标准 ①2014年1月1日至2017年2月1日就诊于我院且确诊EE的患儿;②EEG检查数据完整;③ 确诊EE后或维持原有治疗或调整治疗(以药物治疗为基础,结合生酮饮食治疗和手术治疗);④至少完成一次在我院就诊后≥6个月的随访。
1.3 EE诊断和治疗 图1为我院EE诊断和治疗流程。
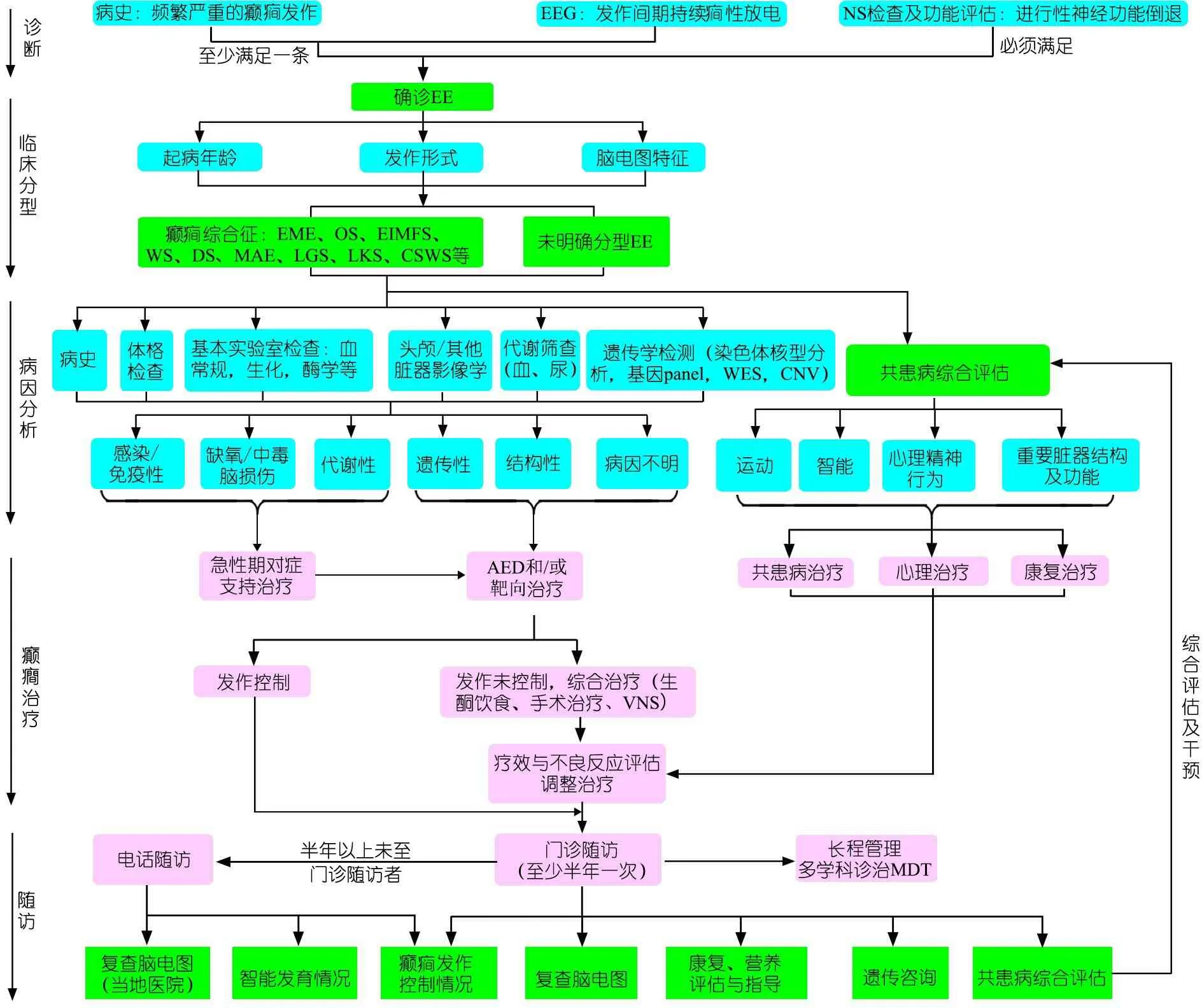
图1癫性脑病诊治流程图
治疗有效率=有效例数(包括发作控制及有效或认知功能恢复至正常和改善)/治疗总例数×100%。
1.7 统计学分析 采用SPSS 20.0软件进行数据分析。基本病例资料采用描述性统计分析;分类资料以百分比表示。
2 结果
2.1 一般情况 符合本文纳入标准的EE 234例,均为汉族,男150例(64.1%),女84例;起病年龄中位数6(0~96)个月,其中3个月以内起病36例(15.4%),1岁以内起病165例(70.5%)。
2.3 体格检查 头围<2 SD 12例,特殊面容6例;神经皮肤综合征5例(结节性硬化4例,神经纤维瘤病1例);合并先天性心脏病17例,合并生殖器畸形(隐睾、小阴茎、尿道下裂)6例,合并消化道畸形(腹股沟斜疝、先天性胆道闭锁)2例,合并先天性喉软骨发育不良3例,多系统发育异常16例(6.8%)。
2.4 辅助检查
2.4.4 遗传学检测 146例行遗传学检测,行染色体核型分析20例均未见异常;5/22例发现致病性拷贝数变异;34/38例SCN1A基因测序发现致病突变;2/3例行STXBP1基因测序发现致病突变;22/43例WES发现致病或可能致病突变;10/20例行基因panel测序发现致病或可能致病突变。
表1 癫综合征临床及脑电图特征
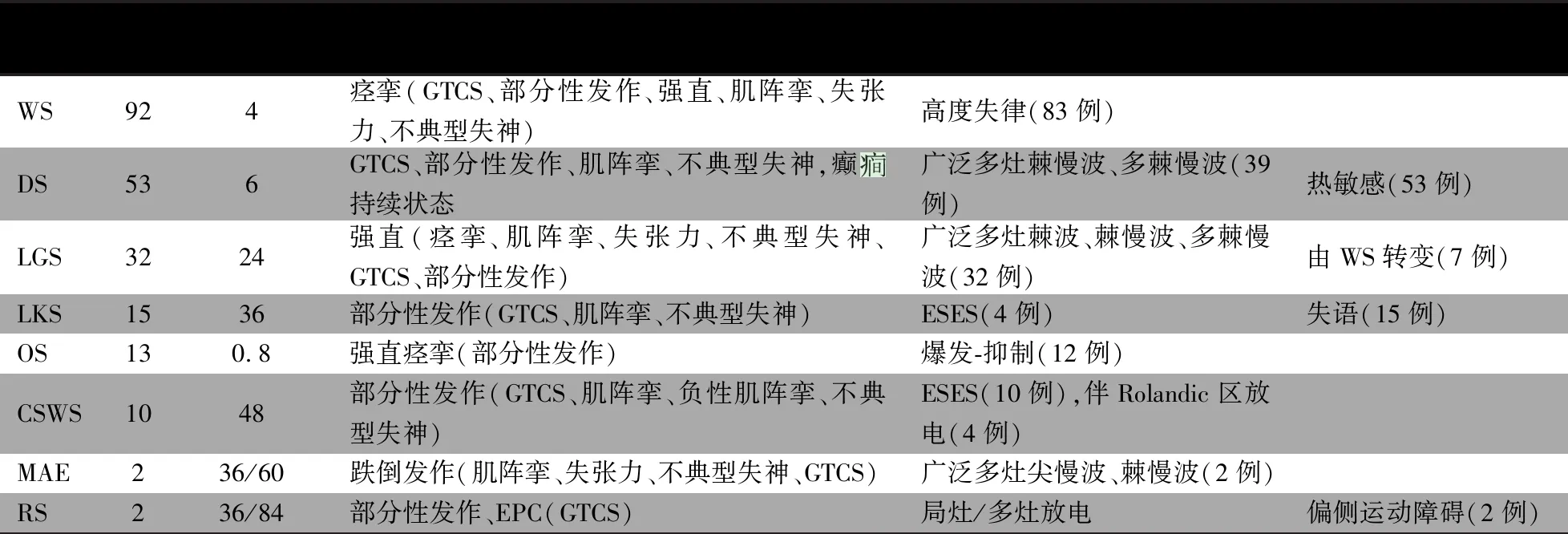
表1 癫综合征临床及脑电图特征
综合征例数起病年龄(中位数,月)主要发作形式(其他发作形式)脑电图特征其他特点WS924痉挛(GTCS、部分性发作、强直、肌阵挛、失张力、不典型失神)高度失律(83例)DS536GTCS、部分性发作、肌阵挛、不典型失神,癫持续状态广泛多灶棘慢波、多棘慢波(39例)热敏感(53例)LGS3224强直(痉挛、肌阵挛、失张力、不典型失神、GTCS、部分性发作)广泛多灶棘波、棘慢波、多棘慢波(32例)由WS转变(7例)LKS1536部分性发作(GTCS、肌阵挛、不典型失神)ESES(4例)失语(15例)OS130.8强直痉挛(部分性发作)爆发-抑制(12例)CSWS1048部分性发作(GTCS、肌阵挛、负性肌阵挛、不典型失神)ESES(10例),伴Rolandic区放电(4例)MAE236/60跌倒发作(肌阵挛、失张力、不典型失神、GTCS)广泛多灶尖慢波、棘慢波(2例)RS236/84部分性发作、EPC(GTCS)局灶/多灶放电偏侧运动障碍(2例)
注 WS:West综合征;DS:Dravet综合征:LGS:Lennox-Gastaut综合征;LKS:Landau-Kleffner综合征;OS:大田原综合征;CSWS:伴睡眠期持续棘慢波的癫性脑病;MAE:肌阵挛-失张力癫;RS:Rasmussen综合征;GTCS:全面性强直-阵挛发作;EPC:持续性部分性癫;ESES:睡眠中癫性电持续状态
表2 各临床分型癫性脑病的已知病因分类[n(%)]
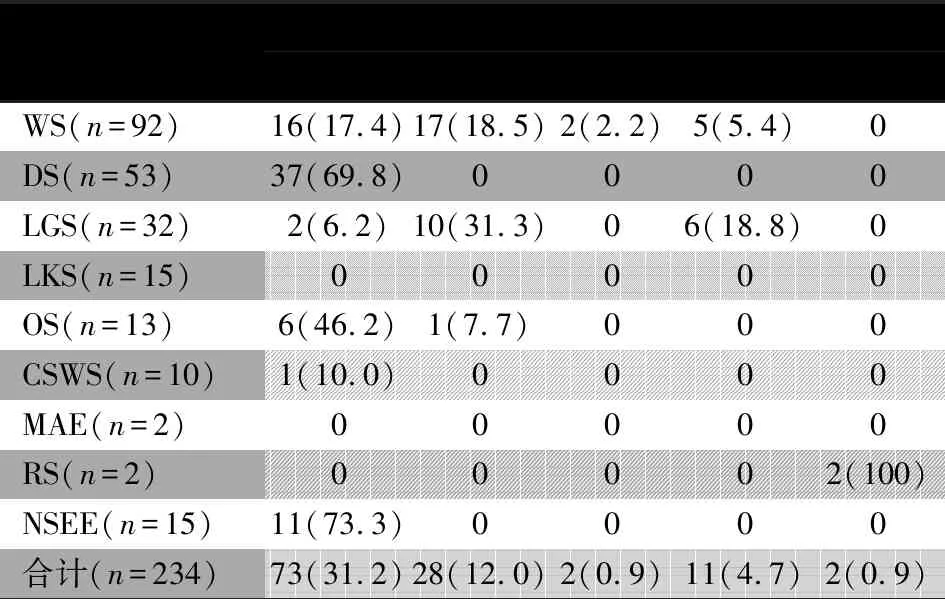
表2 各临床分型癫性脑病的已知病因分类[n(%)]
临床分型已知病因遗传性结构性代谢性感染性免疫性WS(n=92)16(17.4)17(18.5)2(2.2)5(5.4)0DS(n=53)37(69.8)0000LGS(n=32)2(6.2)10(31.3)06(18.8)0LKS(n=15)00000OS(n=13)6(46.2)1(7.7)000CSWS(n=10)1(10.0)0000MAE(n=2)00000RS(n=2)00002(100)NSEE(n=15)11(73.3)0000合计(n=234)73(31.2)28(12.0)2(0.9)11(4.7)2(0.9)
2.7 疗效 ①AED(未接受生酮饮食和手术治疗前):临床发作控制30例(12.8%),治疗有效49例(20.9%),治疗无效155例(66.2%),药物治疗有效率33.7%。②AED+生酮饮食:临床发作控制6例(8.1%),治疗有效29例(39.2%),治疗无效39例(52.7%),有效率47.3%。其中CSWS 5/5例均有效(100%),DS 10/13例有效(76.9%),LKS 2/4例有效(50%),WS 14/29例有效(48.3%)。③AED+手术:3例临床发作控制(分别随访21月、22月、26月无发作),2例无效,手术治疗有效率60.0%。④AED+VNS:4例治疗有效,5例无效,VNS治疗有效率44.4%。
234例综合治疗疗效分析:临床发作控制37例(15.8%),治疗有效77例(32.9%),治疗无效120例(51.3%),综合治疗有效率48.7%。
2.8 随访 近半年内至少完成1次门诊随访127例(54.0%),余均通过电话随访;233例病程≥1年(1例于9月龄死亡),209例病程≥2年,随访病程中位数44(9~211)月,随访年龄中位数54(9~217)月。
2.8.1 发育及共患病评估 发病前智能发育评估,139例(59.4%)发育正常;64例(27.4%)存在发育异常(3例精神发育迟缓,61例精神运动发育迟缓);31例起病年龄<2月,发育情况不详。发病后首次智能发育评估,11例(4.7%)发育正常;223例(95.3%)存在发育异常(24例精神发育迟缓,1例运动发育倒退,198例精神运动发育迟缓),其中64例行DST评估,平均DQ<53,MI<54。治疗后(随访病程中位数44月)智能发育随访,19例(8.1%)发育正常;215例(91.9%)存在发育异常(22例精神发育迟缓,193例精神运动发育迟缓),其中65例行DST评估,平均DQ<52,MI<54,13例行韦氏智测,平均IQ<57。5例通过DSM-V诊断为孤独症,1例诊断为选择性缄默症。54例行视觉诱发电位(VEP)及听觉诱发电位(AEP)检测,VEP:未引出波形1例,波形分化差13例,潜伏期延长、振幅下降4例;AEP:重、中和轻度听力下降分别为1、7和5例。
图2234例癫性脑病患儿抗癫药物选择(例次/百分比)
注 A:West综合征92例;B:Lennox-Gastaut综合征32例;C:大田原综合征13例;D:Dravet综合征53例;E:Landau-Kleffner综合征15例及CSWS 10例;F:其他癫性脑病19例;VPA:丙戊酸钠;TPM:托吡酯;LEV:左乙拉西坦;CLZ:氯硝西泮;NZP:硝西泮;PB:苯巴比妥;OXC:奥卡西平;CBZ:卡马西平;LTG:拉莫三嗪;VGB:氨己烯酸;PDN:泼尼松;ACTH:促肾上腺皮质激素
3 讨论
EE的病因复杂多样,根据2017年ILAE分类建议,可分为遗传性、结构性、代谢性、感染性、免疫性及病因不明6大类[5]。其中遗传因素具有重要作用,基因突变是EE主要的遗传病因[6]。同一病例可有多个病因分类,例如结节性硬化症患者既存在结构性病因,也存在遗传性病因。本文中有5例存在结构性病因和1例存在代谢性病因者行基因检测发现致病突变,其中3例结节性硬化检出TSC2基因突变,1例神经纤维瘤病检出NF1基因突变,1例LGS伴多发多小脑回畸形检出PAFAH1B1基因突变,1例Menkes病检出ATP7A基因突变。
EE的治疗尚面临巨大挑战。目前EE的治疗手段包括AED治疗、生酮饮食治疗及手术治疗等。除了少部分具有特定病因的EE患儿通过早期有针对性的治疗干预能获得较好的疗效及预后,大多数患儿由于缺乏准确的病因诊断,仅为对症性治疗,往往对AED疗效不佳,预后不良[9]。本研究中,234例患儿均接受AED治疗,其中仅12.8%通过AED治疗达到临床发作控制,66.2%治疗无效,提示AED治疗对于多数EE患儿疗效不佳。
