基于TCGA数据集分析线粒体转录终止因子2基因在宫颈癌的表达及意义*
2019-03-22普元倩王唯斯张若鹏
普元倩,梅 雯,王唯斯,李 彬,张 态,张若鹏,熊 伟
宫颈癌是全球妇女中仅次于乳腺癌的第二个最常见的恶性肿瘤。在我国,宫颈癌的每年新发病例为13.2万,约占世界新发病例的28.8%[1]。宫颈癌病死率居女性恶性肿瘤病死率的第二位,在女性生殖器官肿瘤中居首位[2]。肿瘤的生物学特性不仅取决于核内遗传物质,而且与核外的线粒体DNA(mitochondrial DNA,mtDNA)也有一定的关系[3]。线粒体转录终止因子 (mitochondrial transcription termination factor,MTERF)是一类由核基因编码通过特殊的方式转运到线粒体,并能与mtDNA特异结合的蛋白质,在mtDNA的复制、转录以及翻译中发挥调控作用[4]。线粒体转录终止因子蛋白家族有4个成员(MTERF1~F4),其中线粒体转录终止因子2 (mitochondrial transcription termination factor 2,MTERF2)又称线粒体转录终止因子D3(mitochondrial transcription termination domain containing 3,MTERFD3)或线粒体转录终止样因子(mitochondrial transcription termination-like factor,MTERFL)[5]。MTERF2基因是通过比较正常血清培养与血清饥饿培养的人成纤维细胞基因表达谱差异发现的一个细胞增殖抑制基因[6]。人MTERF2基因定位于12q23.3,可编码由385个氨基酸残基构成的蛋白质,定位于线粒体内[7]。研究表明,人MTERF2蛋白在体内与mtDNA的结合没有序列特异性,且抑制线粒体基因的表达[8-11]。MTERF2蛋白在线粒体内的含量相对丰富,是线粒体拟核的重要组成成分[12]。目前,对于MTERF2基因在宫颈癌发生发展的作用及其机制尚不清楚,该研究旨在利用TCGA数据集分析MTERF2在宫颈癌中的表达及其预后意义。
1 材料与方法
1.1 数据集下载 利用R3.3.3软件Bioconductor/TCGA Biolinks函数包从 TCGA 数据库(https://tcga-data.nci.nih.gov/tcga/)下载并预处理宫颈癌数据集的MTERF2 mRNA表达RNA SeqV2数据。
1.2 GEPIA数据库分析TCGA数据集中宫颈癌和正常宫颈组织MTERF2表达差异 基因表达谱动态分析 (Gene Expression Profiling Interactive Analysis,GEPIA)(http://gepia.cancer-pku.cn/) 是由北京大学研制开发可用于分析基因在癌症和正常组织的差异表达的在线应用,可对TCGA数据库进行可视化分析。TCGA宫颈癌数据集中包括306例宫颈癌组织样本和13例正常宫颈组织样本,用GEPIA分析MTERF2在宫颈癌和正常宫颈组织中的表达差异。
1.3 数据集筛选与宫颈癌临床病理学参数资料相关研究 TCGA数据库中下载得到含有人MTERF2 mRNA表达量的宫颈癌组织样本306例,剔除临床病理参数不详或不完整的病例以及重复的资料病例,仅保留TCGA数据集中包含完整临床参数和生存资料的病例,最后得到含有完整临床病理参数和生存资料的病例246例。按病例的ID号将患者的临床资料与MTERF2 mRNA表达RNA SeqV2数据合并成同一个Excel表格。根据 表达谱数据,对宫颈癌组织样本的MTERF2表达量进行由低到高排序,样本中MTERF2表达量为62.420~911.306,中位数为212.666。将数据进行二等分,低于中位数的样本为低表达组,高于中位数的样本为高表达组。获取的TCGA数据集宫颈癌患者MTERF2低表达组123例,高表达组123例。
1.4 统计学方法建立患者临床资料库,将患者年龄、肿瘤大小及浸润深度、淋巴结转移、远处转移、AJCC临床分期、病理分级、病理类型、MTERF2 mRNA表达水平等指标进行量化赋值后进行统计学分析,具体赋值见表1。使用R 3.3.0软件进行统计学分析,采用Shapiro-wilk法检测宫颈癌组织中MTERF2 mRNA表达量的数据正态分布情况,结果表明MTERF2 mRNA表达水平不符合正态分布,所以宫颈癌组织中MTERF2表达水平采用Mannwhitney U非参数检验。临床病理参数相关性分析,组间比较采用χ2检验及Fisher确切概率法。使用R语言Survival软件包以Kaplan-Meier法绘制生存曲线并进行Log-rank检验,多因素分析采用Cox回归模型。以P<0.05为差异有统计学意义。
2 结果
2.1 宫颈癌与正常宫颈组织MTERF2表达差异使用GEPIA数据库对TCGA数据集进行数据挖掘和可视化分析,结果发现,相较于正常宫颈组织,宫颈癌组织中MTERF2 mRNA表达水平显著降低,且差异具有统计学意义(P<0.05)。见图1。

图1 宫颈癌组织和正常宫颈癌组织中MTERF2 mRNA 表达差异(*P<0.05)
2.2 MTERF2表达与宫颈癌患者临床病理学参数相关性 在TCGA数据集中共纳入246例临床病理参数完整的病例资料,年龄21~85岁,中位年龄46岁。对该数据集中宫颈癌患者预后影响因素进行赋值(表1),通过统计分析资料可知,宫颈癌患者MTERF2 mRNA表达量与肿瘤浸润深度、淋巴结转移、远处转移、AJCC临床分期和病理分级无关(P>0.05);与年龄(P=0.018)、病理类型(P<0.001)明显相关。见表2。

表1 宫颈癌患者预后影响因素及其赋值

图2 人MTERF2 mRNA表达水平与宫颈癌患者预后的相关性

表 2MTERF2 mRNA表达水平与宫颈癌临床病理参数的相关性分析(n=246)
2.3 MTERF2表达与宫颈癌患者预后的相关性使用R语言Survival软件包对TCGA数据集中MTERF2高表达组和MTERF2低表达组宫颈癌患者的预后进行Kaplan-Meier分析和Log-rank检验,分别绘制总体生存率(overall survival,OS)和无疾病进展生存率(disease free survival,DFS)生存函数曲线(图2)。结果表明,MTERF2 mRNA表达量与宫颈癌患者的OS和DFS均无明显的相关性(Log-rank P>0.05)。
2.4 影响宫颈癌患者预后的COX多因素回归分析 单因素分析结果显示,肿瘤浸润程度、淋巴结转移、远处转移、AJCC临床分期均能影响患者的预后(P<0.05)。 年龄、病理分级、病理类型、MTERF2 mRNA表达量与宫颈癌患者预后无相关性 (P>0.05)。将单因素分析中对预后有影响的指标纳入COX模型行多因素回归分析,结果提示,淋巴结转移是影响宫颈癌患者预后的独立因素(P<0.05)。见表3。
3 讨论
癌症的发生不仅与核内遗传物质相关,还与核外的mtDNA密切相关。由于mtDNA缺乏组蛋白的保护,无DNA修复系统,处在高氧化应激环境中,容易受电子传递链产生的自由基的损伤,与化学致癌物的亲和力较核基因组高等原因,使mtDNA具有较高的突变率,可能具有潜在的致癌性[13]。研究发现,在多种癌组织及其相应的体液标本存在mtDNA的突变,如宫颈癌、卵巢癌、头颈癌、膀胱癌和大肠癌等[14-17]。恶性肿瘤的发生不仅与mtDNA结构改变相关,还与其数量有密切联系。与正常组织相比,不同类型的肿瘤组织中mtDNA数量有着不同程度的增多或者减少[18]。线粒体是细胞内重要的细胞器,线粒体功能障碍与恶性肿瘤、线粒体糖尿病、神经退行性疾病等疾病的发生密切相关[19]。研究表明,细胞内过表达MTERF2基因显著抑制细胞生长和增殖的作用,细胞周期阻滞于G1/S期,但并不诱导细胞凋亡[6]。最近,Han等报道人MTERF2蛋白对1-甲基-4苯基吡啶离子 (1-methyl-4-phenyl-pyridiniuiodide,MPP+)诱导的神经细胞线粒体功能障碍和细胞损伤具有促进作用[20]。MTERF2是调控线粒体基因表达的重要蛋白因子,目前,MTERF2在肿瘤中的表达及其预后意义目前尚未见报道。
该研究中,笔者使用R语言软件从TCGA数据库下载306例宫颈癌组织和13例正常宫颈组织RNA SeqV2数据。GEPIA分析结果发现,相较于正常宫颈组织,宫颈癌组织中MTERF2 mRNA表达水平显著降低,且差异具有统计学意义(P<0.05),结果提示MTERF2基因在细胞内的功能可能类似于细胞抑癌基因。TCGA数据集处理后获取246例宫颈癌患者MTERF2 mRNA表达量、临床病理资料等完整的数据,根据MTERF2 mRNA的中位表达量,将研究对象分为MTERF2高表达及低表达两组。分析发现,宫颈癌患者MTERF2 mRNA表达量与肿瘤浸润深度、淋巴结转移、远处转移、AJCC临床分期、病理分级均无关(P>0.05);而与年龄、病理类型相关(P<0.05)。进一步Kaplan-Meier生存分析宫颈癌患者MTERF2表达量与预后的关系,结果提示MTERF2 mRNA高表达组与低表达组的总体生存率及无疾病进展生存率的差异无统计学意义 (P>0.05)。单因素分析显示,肿瘤浸润深度、淋巴结转移、远处转移、临床分期均能影响患者的预后(P<0.05)。年龄、病理分级、病理类型、MTERF2 mRNA表达与宫颈癌患者预后无相关性(P>0.05)。COX多因素回归分析结果提示,淋巴结转移是影响宫颈癌患者预后的独立因素(P<0.05)。
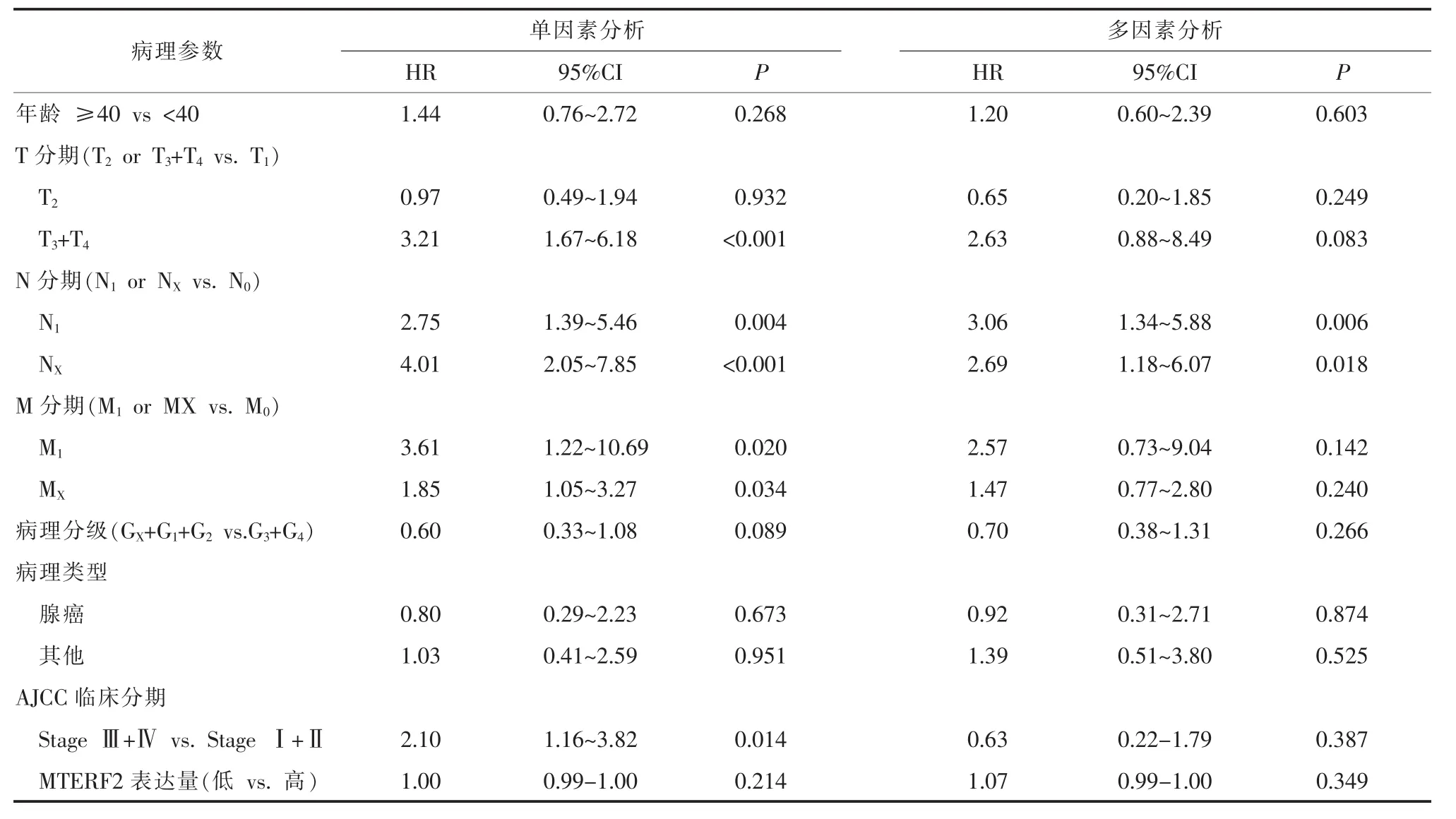
表3 影响宫颈癌患者预后的单因素和多因素回归分析
该研究的优势在于利用TCGA在线数据库,样本量较大,临床资料完整。而不足之处在于,TCGA数据集中提供的是mRNA水平的表达数据,可能无法完全代表MTERF2在蛋白质水平的表达情况。在后续的研究中应该结合Western blot和免疫组织化学方法进一步分析讨论。该研究为MTERF2在肿瘤中的基础和临床研究提供了线索和依据,需要进一步的研究更深入地探讨MTERF2在肿瘤发生发展中的作用及机制,为肿瘤的靶向治疗提供新思路。
