密度泛函理论研究有机太阳能电池界面的激子分离及电荷转移速率:DR3TBDT/PC60BM 体系
2019-03-19王冬梅
王冬梅, 田 军,2, 郑 茹, 柴 垚
(1.宝鸡文理学院化学化工学院, 宝鸡 721013; 2.岐山县青化镇初级中学, 宝鸡 722402)
1 引 言
目前,最普通的硅太阳能电池能量转换效率power conversion efficiency(PCE)已经达到~30%的理论值[1], 然而高昂的生产成本阻碍了其广泛的应用.而有机太阳能电池以其低廉的价格、环境友好、简单的制作工艺等优势在近些年吸引着广泛的关注,并有望成为硅光伏的替代品.
有机太阳能电池器件通常具有三明治的夹心结构,即,给受体材料混合后被夹在两个电极中间,其中阳极是透明的,透过太阳光.有机太阳能电池的光电转换效率主要受以下五个步骤的控制[2, 3]:(1)活性材料吸收太阳光并产生激子(光吸收效率ηA)(2)激子扩散直到D/A界面(扩散效率ηD),(3)激子在异质结界面处分离并伴随电荷转移态Charge transfer(CT)的产生(分离效率ηCS),(4)电荷(空穴和电子)分别在给受体材料中传输并到达电极后富集(收集效率ηCC).在整个光致电荷产生的过程中,由(3)到(4),电子和空穴也会发生重组(重组效率ηCR)又回到基态,因此,要提高器件的整体效率,ηA,ηD,ηCS和ηCC应该最大化,而ηCR应该最小化.然而,目前在有机太阳能电池中很难建立起过程中的每种效率与界面化学结构之间的关系,并且对于光伏活性材料的设计没有简单准则去校正能级并预测材料应有的加工过程.幸运的是,我们可以预测发生在电池中基本过程的速率.这些过程,如光吸收,激子形成以及电荷迁移,它们仅发生在异质结材料中而不包括界面.电荷分离(charge separation(CS))和电荷重组(charge recombination(CR))是两个关键的界面过程,它们的竞争直接影响电池效率.预测这两个过程的速率是估计界面电荷产生效率的第一步,从而指导新材料的设计.然而,估计这两个过程的速率面临最大的挑战就是,它们的计算受到诸多因素的影响,如计算方法和模型的选择等.尽管如此,我们在处理同一系列的分子时,选择相同的计算方法和模型就可以定性地比较速率的大小趋势,从而对材料进行筛选.
鉴于当前有机太阳能电池中光活性材料的理论研究大多数只停留在给受体材料的设计以及相关光谱性质的计算等方面,而有关给受体材料界面电荷转移的理论研究相对较少且不成熟.电池效率方面,虽然最好的有机光伏器件效率已经超过10%[4],但离商业化生产仍有一定的距离.为了进一步从材料分子的角度改善器件整体效率,本文以光伏活性材料中最具代表性的给体材料聚-3己基噻吩(P3HT)与受体材料苯基丁酸甲酯(PC60BM)组成的异质结体系[5-7]为参照,选取近期在实验中合成的经典给体材料分子DR3TBDT,模拟该分子与经典受体材料PC60BM形成的DR3TBDT/PC60BM复合体系[8],并以此为模型(图1),从动力学角度深入研究了给受体界面的电荷分离情况.在理论研究过程中,通过采用量子化学中的密度泛函理论方法,分别计算了孤立的给受体分子以及复合物的基态结构性质、吸收性质、激发态电荷转移,并通过Rehm-Well表达式[9],Marcus理论的双势阱[10]、双球棍模型[11]以及广义的Mulliken-Hush (GMH) 模型[12]分别计算了电子转移和电荷重组过程中的Gibbs自由能变、内外重组能以及电子耦合,最后通过Marcus电荷转移速率方程得出了界面的电荷转移和重组速率,从而为新材料的设计从动力学角度提供理论表征手段.
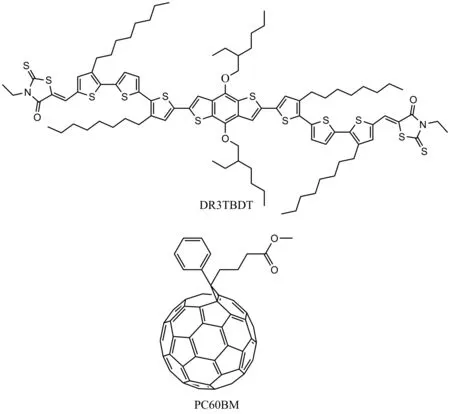
图1 DR3TBDT和PC60BM的结构Fig.1 The structure of DR3TBDT and PC60BM
2 理论框架
2.1 计算细节
由于给受体间存在弱相互作用,DR3TBDT/PC60BM体系的基态构型优化采用了密度泛函理论中考虑了色散校正的ωB97X-D泛函和6-31G(d)基组,为保证计算结果的一致性,孤立的给受体分子DR3TBDT和PC60BM也采用相同的计算水平进行优化.所有分子的光谱吸收以及有关激发态的计算均采用含时密度泛函方法中的长程相关泛函CAM-B3LYP[13]和6-31G(d)基组[14]. Gibbs自由能、重组能和转移积分的计算中,给受体分子相应态的能量计算均在B3LYP/6-31G(d)水平上完成.由于侧链只是用来增加溶解性而不影响电子性质[15, 16],因此为了节省机时,DR3TBDT中长的烷基链和烷氧基链都用H代替.所有计算都是在Gaussian09程序包中完成.此外,态密度图、电荷差分密度图均采用Mutiwfn 3.1程序完成.
2.2 界面模型
给受体分子之间最初的相对位置对复合物优化的结果以及界面电荷转移和电荷重组速率常数的估计都有着显著的影响.2009年,Bredas等人研究了并五苯/富勒烯体系给受体的两种初始相对位置(并五苯平行或垂直于受体)对激子分离和电荷重组过程的影响[17].2011年,Alessandro等人研究了P3HT6/PC60BM体系给受体的八种初始相对位置[18]对界面电荷转移和重组绝对速率的影响.研究表明,给受体初始不同位置对激子分离过程中的电子耦合影响最为显著.在本文中,我们采用了最有利于分子间电子耦合的初始位置,即,PC60BM的两个六边形公用的棱与给体分子长轴方向所在的平面平行,如图2所示,点a与b之间的初始距离为3.5 Å[19].从孤立分子到复合物,分子间的相互作用能可表示为:
△E=ED/A-(ED+EA)
(1)
式中,ED、EA和ED/A分别是给体、受体和复合物基态的能量.
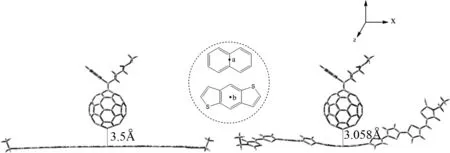
图2 复合物DR3TBDT/PC60BM的初始构型(左)和优化后的构型(右)Fig. 2 The original model(left) and optimized(right) (ωB97XD/6-31G(d)) structure for DR3TBDT/PC60BM
2.3 Marcus速率方程
Marcus理论被广泛地用于描述包括有机太阳能电池在内的给-受体体系的电荷转移和电荷重组动力学[20].
(2)
式中,λ是重组能,VDA是给受体之间电子耦合,也叫电荷转移积分.ΔG是电子转移反应的吉布斯自由能变,kB是玻尔兹曼常数,h是普朗克常数,T是温度(通常按300 K计算).通过计算λ、VDA和ΔG这三个参数就可以得到电荷转移和重组速率常数.
3 结果与讨论
3.1 基态性质
基于给定的模型,我们优化了复合物DR3TBDT/PC60BM,发现优化前后分子构型发生了明显变化,给体分子由原来的平面构型变成两端扭曲的结构,如图2.根据式(1),我们计算了孤立分子形成复合物的结合能,见表1.可以看出,它们之间的结合能在分子间作用力的范围内(几个千焦到几十个千焦),是一种弱相互作用[21].
表1 在ωB97X-D/6-31G(d)水平上计算的DR3TBDT, PC60BM和DR3TBDT/PC60BM的能级、稳定化能及结合能
Table 1 The calculated (ωB97XD/6-31G(d)) energy levels and stabilization energies of DR3TBDT, PC60BM and complex(DR3TBDT/PC60BM) as well as the binding energies between the isolate molecules and complex
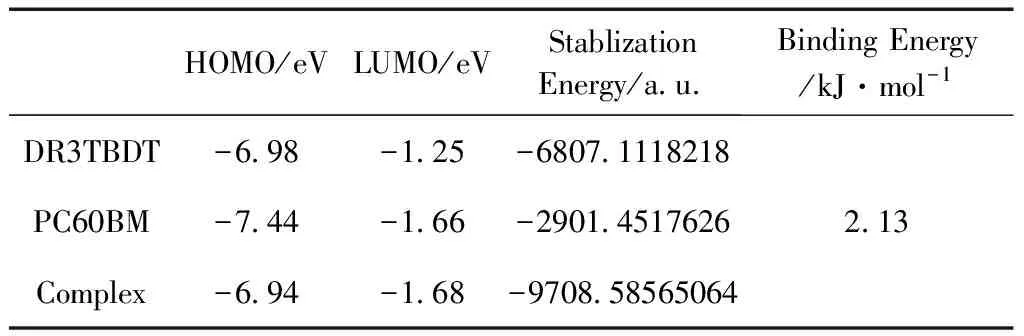
HOMO/eVLUMO/eVStablizationEnergy/a.u.Binding Energy/kJ·mol-1DR3TBDT-6.98-1.25-6807.1118218PC60BM-7.44-1.66-2901.4517626Complex-6.94-1.68-9708.585650642.13
注:表中HOMO为Highest Occupied Molecular Orbital的缩写,LUMO为Lowest Unoccupied Molecular Orbital的缩写.
此外,我们还计算了分子总的态密度density of states(DOS)和部分态密度partial density of states(PDOS),如图3所示.(a)和(b)图分别是DR3TBDT的态密度图,可以看出,侧链C8H17和OC8H17对DR3TBDT分子轨道的贡献很小,因此,通过简化模型对后续工作的研究是可信的.(c)和(d)图分别是DR3TBDT和PC60BM对复合物分子轨道贡献的态密度图.从图中可以看出,对于复合物而言,电子密度在HOMO和LUMO的分布范围为:-6.98~-7.02 eV(-0.258 a.u.~-0.062 a.u.),对于DR3TBDT,电子密度在HOMO和LUMO 上的分布范围为:-6.98~-1.25 eV(-0.257 a.u.~-0.046 a.u.),对于PC60BM,电子密度在HOMO和LUMO上的分布范围为:-7.44~-1.66 eV(-0.273 a.u.~-0.061 a.u.).从单个分子到复合物,HOMO上电子的贡献主要来自给体,而LUMO主要来自受体.并且复合物的带隙与孤立的给受体分子相比,下降了0.39-0.44 eV,这是给受体间电子耦合作用所致.能级如图4所示.
3.2 光谱吸收和电子跃迁
所有分子光谱吸收过程中的主要的电子跃迁、振子强度和轨道贡献均列于表2、3中,模拟的吸收曲线见图5.结合表2和图5(a)可以看出,DR3TBDT有三个吸收峰,最大吸收峰在451 nm处,主要来自HOMO(H)→LUMO(L)和H-1→L+1的S0→S1跃迁,另外两个肩峰在430~370 nm附近,分别来H-1→L和H→L+1的S0→S2跃迁以及H→L+2的S0→S5跃迁.从表3和图5(b)可知,PC60BM的强吸收主要集中在紫外区250-300 nm处,主要来自电子跃迁S0→S26,S0→S34,S0→S35,S0→S36以及S0→S46.并且第一激发态对应的513 nm为弱吸收.计算的50个态的电子跃迁和振子强度列于表3.由表3和图5(c)可知,DR3TBDT/PC60BM的最大吸收峰在502 nm处,为S0→S1的跃迁,最强的吸收峰位于449 nm处,为S0→S6的跃迁,最强吸收峰的位置和曲线的形状与图5(a)相似,这说明复合物对太阳光的吸收主要来自给体分子.
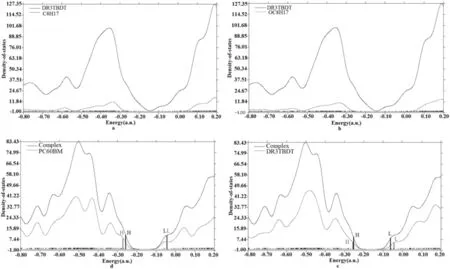
图3 DR3TBDT和DR3TBDT/PC60BM的态密度(DOS)图Fig. 3 Partialdensities of state (PDOSs) for DR3TBDT and complex(DR3TBDT/PC60BM)
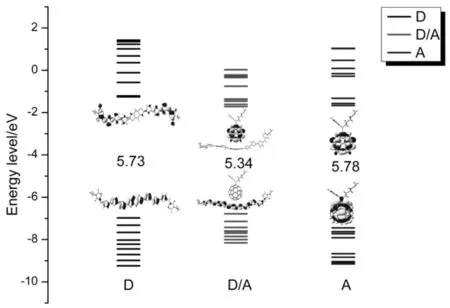
图4 在ωB97X-D/6-31G(d)水平上计算的DR3TBDT(D), PC60BM(A) 和DR3TBDT/PC60BM(D/A)的前线分子轨道能级Fig. 4 The frontier molecular orbital energy levels for DR3TBDT(D), PC60BM(A) and Complex(D/A)
表2 在C-PCM(CHCl3)-CAM-B3LYP/6-31G(d)水平上计算的DR3TBDT跃迁能(eV, nm),振子强度(f)和轨道贡献
Table 2 The calculated(C-PCM(CHCl3)-CAM-B3LYP/6-31G(d)) transition energies(eV, nm),oscillator strengths(f) and orbital contributions for DR3TBDT

E/eV(nm)fOrbital contributionsS12.7487(451.1)4.2659HOMO-1→LUMO+1(31%), HOMO→LUMO (39%)S22.8514(434.8)0.1980HOMO-1→LUMO(41%), HOMO→LUMO+1 (31%)S53.3232(373.1)0.7000HOMO→LUMO+2 (47%)
3.3 激发态电荷转移
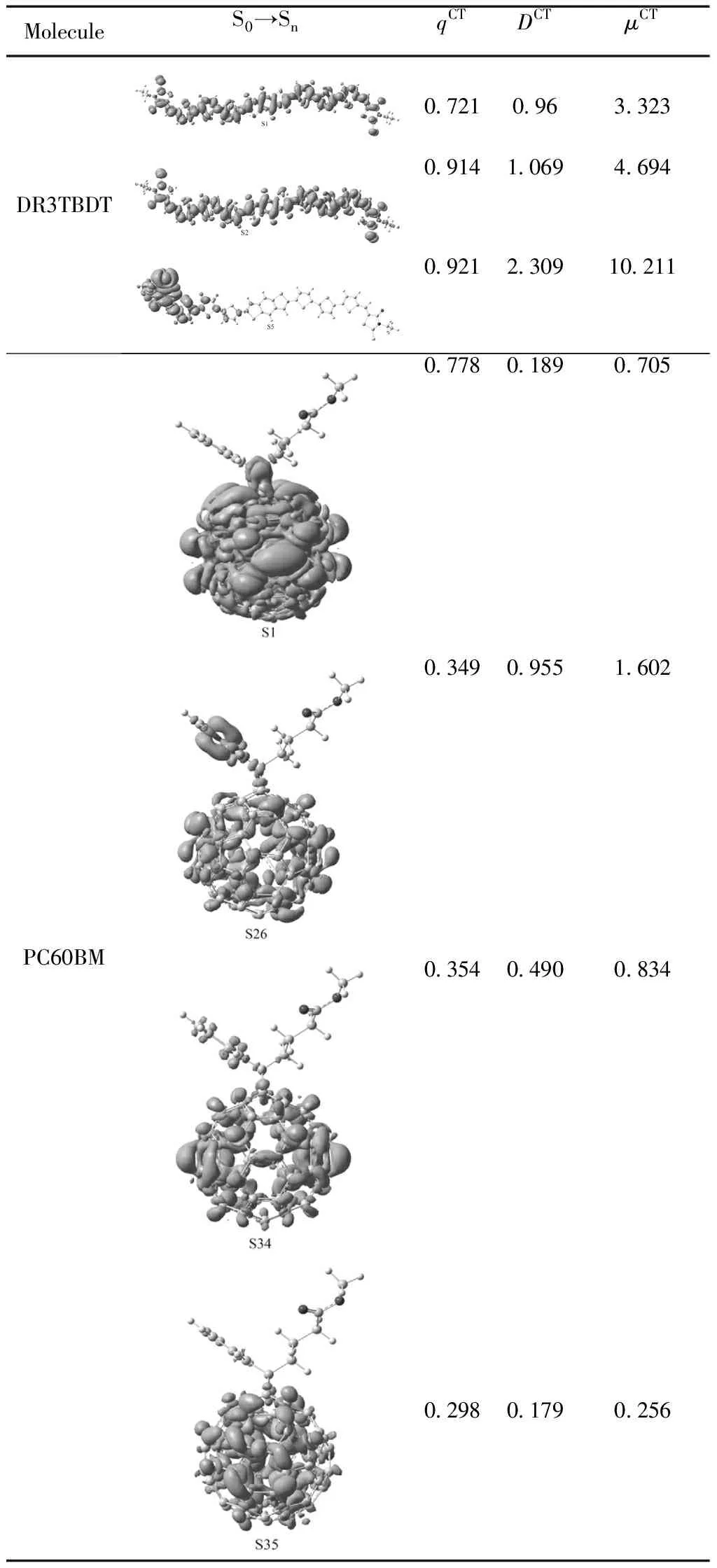
为了清楚地阐明从基态到激发态的电荷转移以及激发态的电荷分布,我们绘制了孤立分子和复合物分子基于S0→Sn跃迁的电荷密度差分图charge density difference(CDD)并计算了电荷转移(CT)参数(DCT,qCT和μCT),如表4所示.其中,DCT是激发过程中电子密度增加的区域和密度减小的区域的重心距离,qCT是电荷转移数目,μCT是变化的偶极距.可以看出DR3TBDT在从S0分别到S1和S2时,电子密度变化发生在整个分子骨架上,从电荷转移参数值看,S1和S2相差不大,说明它们有相似的电荷转移特征.而S0到S5跃迁时,
表3 在C-PCM(CHCl3)-CAM-B3LYP/6-31G(d)水平上计算的PC60BM和复合物DR3TBDT/PC60BM(D/A)的跃迁能(eV, nm)及振子强度(f)
Table 3 The calculated(C-PCM(CHCl3)-CAM-B3LYP/6-31G(d)) transition energies(eV, nm) and oscillator strengths(f) for PC60BM and Complex

PC60BME/eV(nm)fComplexE/eV(nm)fS12.4145(513.5)0.00392.4654(502.8)0.0030S22.4467(506.8)0.00002.5035(495.2)0.0000S32.5289(490.3)0.00002.5880(479.0)0.0000S42.5475(486.7)0.00002.5970(477.4)0.0000S52.6774(463.1)0.00032.7397(452.5)0.0003S62.7242(455.1)0.00052.7576(449.6)4.1894S72.7850(445.2)0.00002.7792(446.1)0.0011S82.8280(438.4)0.00002.8386(436.8)0.0719S92.8702(431.9)0.00162.8403(436.5)0.0313S102.9555(419.5)0.00232.8633(433.0)0.0006S112.9723(417.1)0.00222.8849(429.7)0.0016S122.9958(413.9)0.00003.0105(411.8)0.0024S133.0876(401.6)0.00013.0489(406.6)0.0005S143.1388(395.0)0.00383.0710(403.7)0.0000S153.1803(389.9)0.01983.0886(401.4)0.0021S163.4683(357.4)0.00363.1390(394.9)0.0059S173.5126(352.9)0.00003.1770(390.2)0.0008S183.5485(349.4)0.00223.2016(387.2)0.0104S193.7122(333.9)0.00663.2238(384.5)0.0016S203.7223(333.0)0.01823.3153(373.9)0.1824S213.7266(332.7)0.0008S223.7890(327.2)0.0025S233.8057(325.7)0.0000S243.8679(320.5)0.0183S253.9027(317.6)0.0002S264.0291(307.7)0.1924S274.0732(304.3)0.0379S284.1200(300.9)0.0057S294.1503(298.7)0.0047S304.1680(297.4)0.0542S314.1950(295.5)0.0004S324.2451(292.0)0.0117S334.2544(291.4)0.0301S344.3315(286.2)0.5654S354.3498(285.0)0.1143S364.3724(283.5)0.2183S374.3951(282.0)0.0133续表3S384.4271(280.0)0.0313S394.4293(279.9)0.0017S404.4997(275.5)0.0861S414.5145(274.6)0.0024
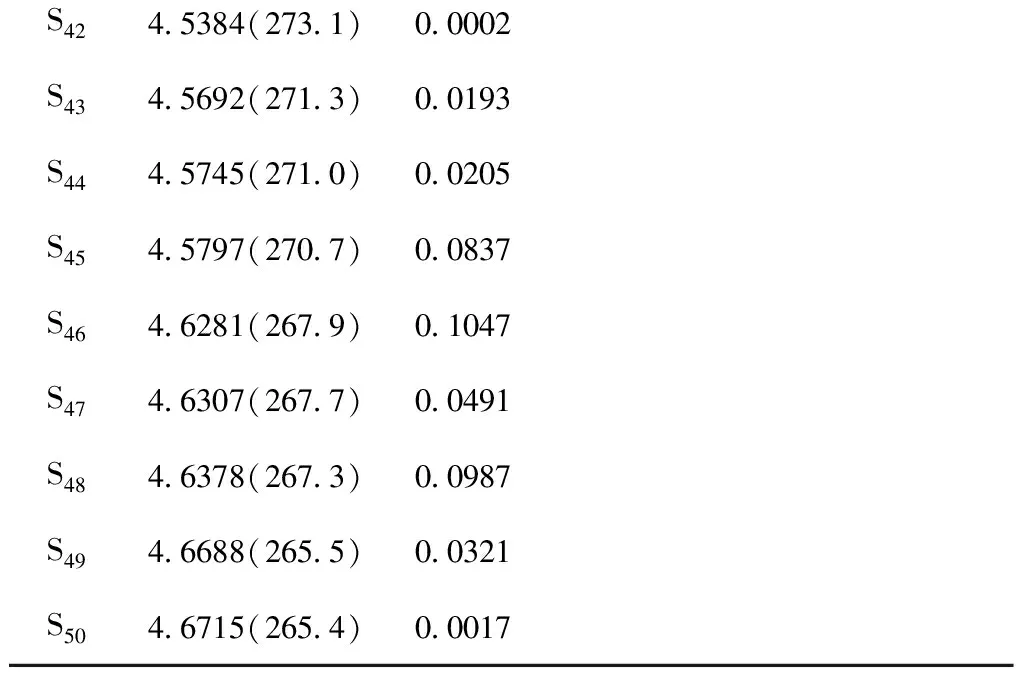
S424.5384(273.1)0.0002S434.5692(271.3)0.0193S444.5745(271.0)0.0205S454.5797(270.7)0.0837S464.6281(267.9)0.1047S474.6307(267.7)0.0491S484.6378(267.3)0.0987S494.6688(265.5)0.0321S504.6715(265.4)0.0017
电荷只集中在了一侧的桥和端基上,在中心给体核和另一侧几乎没有分布.从电荷转移数目来看,从基态到S1、S2和S5很接近,然而随着激发态能级的增加,qCT和μCT都呈现增加的趋势,尤其是S5态的μCT为10.211,说明高激发态下,正负电荷极易发生分离,电子很容易易从DR3TBDT的LUMO转移到PC60BM的LUMO上.而对于孤立的PC60BM,S1态时电荷转移数目最多,而其它几个高能激发态的qCT比较接近,且C60上电荷分布明显减少.S1和S35电荷类似地分布在C60上,而S26,S36,S46电荷在取代基苯环上呈现较多的分布,同时电荷由苯环向C60转移.此外,随着激发态能级的增加,qCT和μCT大致呈增加的趋势,这与线性给体分子中的电荷分布非常相似,即,高激态下激子易解离.对于复合物分子而言,S1和S2呈现明显的电荷转移特征,但是空穴和电子并没有完全地分别分布在给体和受体上,所以S1和S2是分子间电荷转移激发态.
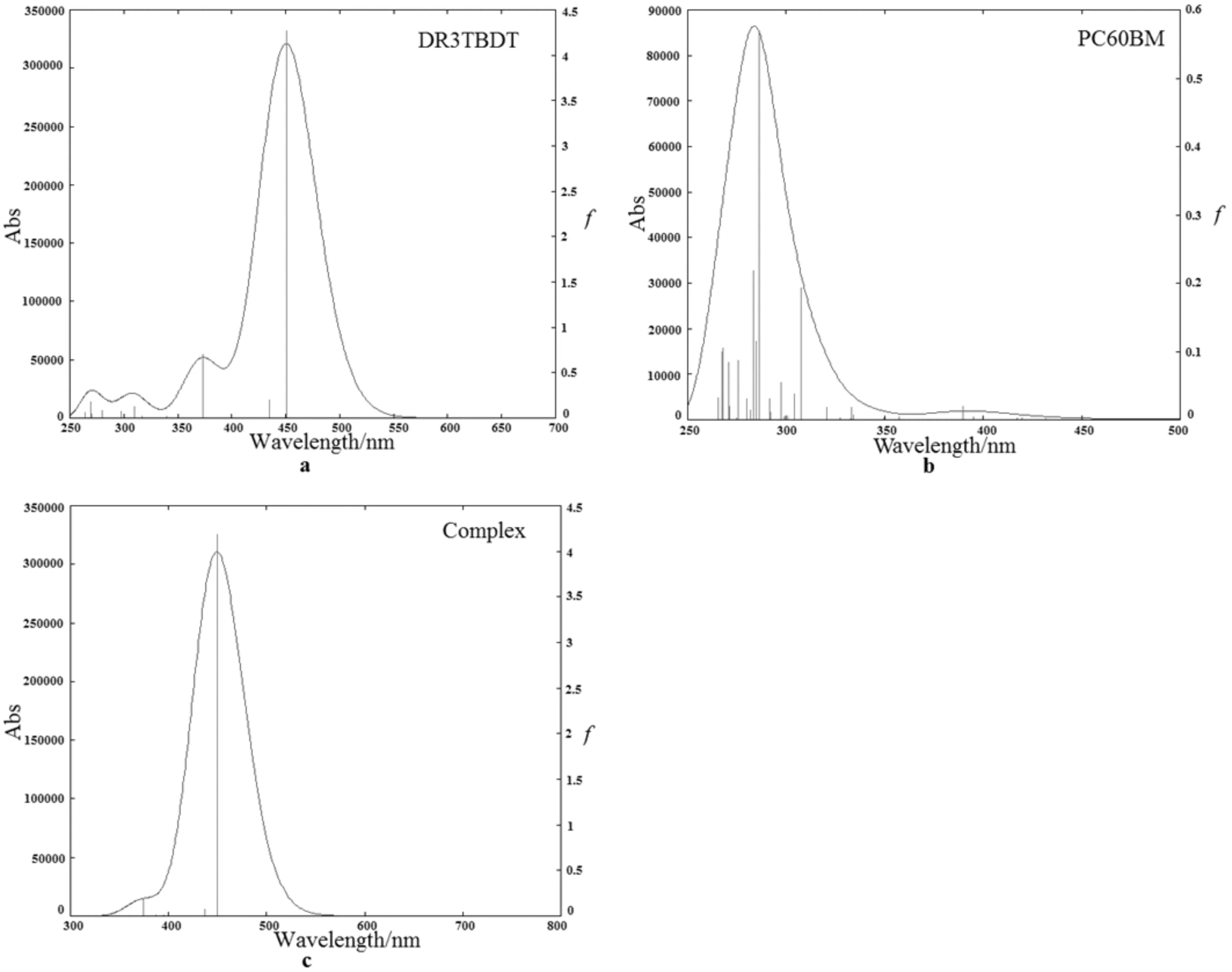
图5 在C-PCM(CHCl3)-CAM-B3LYP/6-31G(d)水平上模拟的DR3TBDT(a)、PC60BM(b)和DR3TBDT/PC60BM(c)的高斯吸收曲线Fig.5 The simulited(C-PCM(CHCl3)-CAM-B3LYP/6-31G(d)) spectra absorption curves of DR3TBDT, PC60BM and Complex
为了获得有效的电荷转移和减缓电荷重组,根据Marcus理论,电荷转移过程中给受体间电子耦合应该最大化,重组能应该最小化.通过计算电荷转移和重组这两个过程中的吉布斯自由能变、重组能以及电子耦合,就可以得到相应速率.
3.4 Gibbs自由能
在激子分离和电荷重组过程中,ΔG分别表示为ΔGCT和ΔGCR.它们的计算可以通过两种方法:一是孤立组分的中性/激发态和阳离子/阴离子态与库仑引力项ΔEC的加和[9],也称为Rehm-Well表达式;另一种方法是分别计算D/A复合物在DA、D*A和D+A-态的能量而求得[18].这两种方式中能量都是通过相应的平衡态构型获得.第一种方法忽略了相互极化效应.第二种方法考虑了极化效应但是需要优化D/A复合物的激发态构型.值得注意的是,不能把ΔGCT简单看作是给受体的LUMO能差,这样会过高估计CT激子的能量.在本文中采用第一种方法来计算ΔGCT和ΔGCR.即:
ΔGCT=E(D+)+E(A-)-E(D*)-E(A)+ΔEC
ΔGCR=E(D)+E(A)-E(D+)-E(A-)+ΔEC
(3)
在吉布斯自由能的计算中,qA和qD分别为给受体相应态的Mulliken电荷,εs是周围溶剂的介电常数,实验中为氯仿(4.80).根据公式(2),可以分别得到ΔGCT和ΔGCR,如表5所示.
3.5 重组能
重组能通常分为内重组能(分子在电子得失过程中几何构型发生变化所引起的)和外重组能(电荷转移过程中周围环境所引起的).内重组能通常采用双势阱模型计算(也称四点法,如图7)[22, 23],用λi表示.
(4)
(5)
图3描述的是D*A和D+A-两个势能面,上述公式中能量E的上标代表反应物所在的态,下标表示态所在的平衡几何构型.
外重组能的精确计算比较复杂.通常使用的是基于Marcus双球棍模型的连续介质模型[11].
表4 DR3TBDT、PC60BM和DR3TBDT/PC60BM基于S0→Sn的电荷密度差分图以及激发态电荷转移数目(qCT/|e-|)、距离(DCT/ Å)和偶极距(μCT/ Debye),紫色和蓝色分别表示电子密度的增加和减少
Table 4 Plotted electronic density difference maps and computed charge transfer distances, amounts and dipoles (DCT,qCTandμCTin Å ,|e-|and Debye, respectively) between S0and Snof all the molecules at the B3LYP/6-31G(d) level. Blue and purple colors correspond to a decrease and increase of electron density, respectively.
MoleculeS0→SnqCTDCTμCTDR3TBDT0.7210.963.3230.9141.0694.6940.9212.30910.211PC60BM0.7780.1890.7050.3490.9551.6020.3540.4900.8340.2980.1790.256
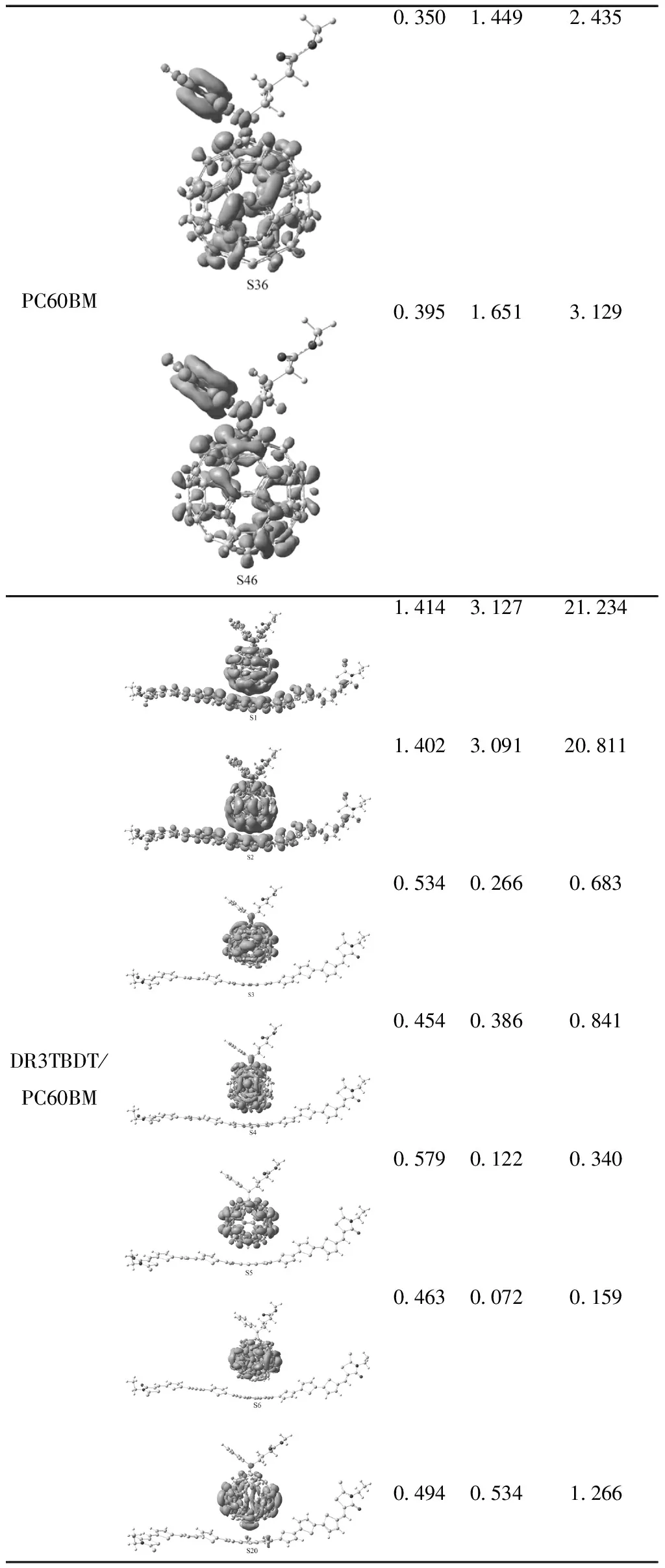
PC60BM0.3501.4492.4350.3951.6513.129DR3TBDT/PC60BM1.4143.12721.2341.4023.09120.8110.5340.2660.6830.4540.3860.8410.5790.1220.3400.4630.0720.1590.4940.5341.266
(6)
式中,dD、dA和dDA分别是给受体半径和给受体的质心距离.εop和εo分别是周围环境的光学介电常数和真空介电常数.
在重组能的计算中[24, 25],我们采用经典的势能面曲线法计算了内重组能,即公式(4),而对于外重组能的计算,涉及到给受体半径和周围溶剂的介电常数.通常把给受体分子看成是带电的球形电荷,采用Gaussian 09中的PCM模型先计算出分子球的体积,从而得到分子半径.根据公式(5),可得外重组能.值得注意的是,电子转移和电荷重组在氯仿溶剂中几乎是同时进行的,所以在计算重组能的时候,需要将每个过程中的内重组能分别与外重组能进行加和.如表6所示.

表5 B3LYP/6-31G*水平上与Gibbs自由能有关的参数
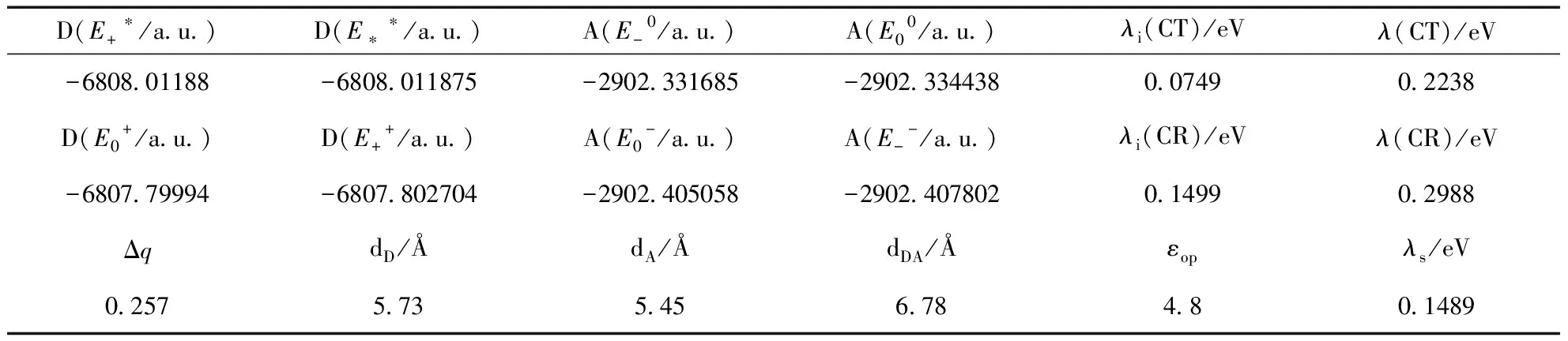
表6 B3LYP/6-31G*水平上与重组能有关的参数
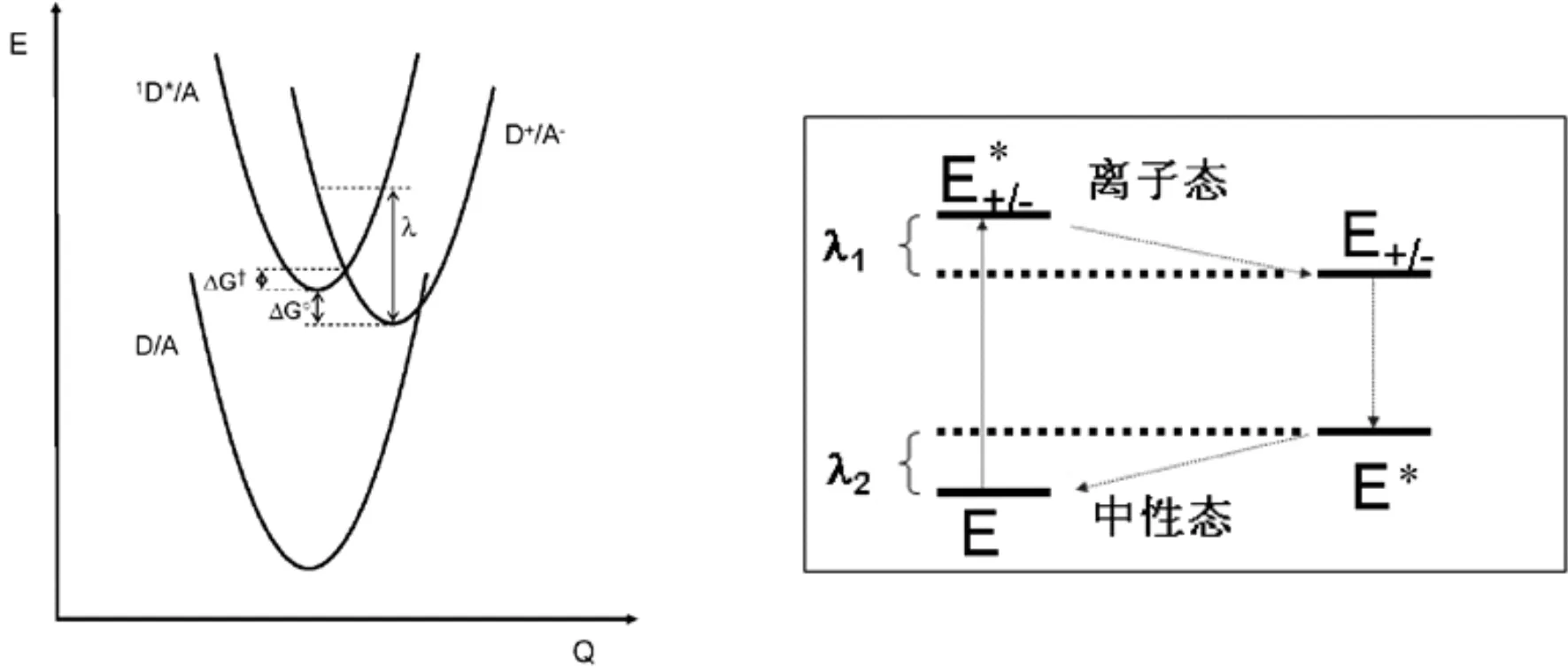
图6 双势阱模型下的内重组能计算方法Fig. 6 Calculation method of internal recombination energy in double-well model
3.6 电子耦合
与重组能的计算相比,目前电子耦合还没有一个完全精确的估计方法,在本文中,我们采用近似的广义Mulliken-Hush(GMH)模型[26, 27]来计算激发态的电荷转移积分,(电子转移耦合矩阵元),这被Sun等人证实很适合我们研究的体系[12].转移积分在两态公式中(S0和Sn态)可表示为:
(7)
式中,μtr是复合物Sn态沿Y轴方向的跃迁偶极距,Δμ是两态的偶极距差,ΔE是垂直激发能.
根据激发态电荷转移的结果,S1和S2是分子间电荷转移激发态,即电子和空穴不完全分布在给体或者受体分子上,所以S1和S2是两个简并的分子间电荷转移激发态.VDA的值等于VDA(S1←S0)和VDA(S2←S0)的加和,表7中的数据结合计算的第一垂直激发能Es1和第二垂直激发能Es2带入根据公式(2)可以得到VDA=55.5 meV.
表7 DR3TBDT/PC60BM基于S0、S1和S2的偶极距以及S0→S1和S0→S2的跃迁偶极距 (单位: Debye)
Table 7 The diploe moments for S0, S1and S2as well as the transition dipole moments for S0→S1(unit: Debye)

μxμyμzμtotS01.99441.2816-1.24862.6794S1-4.1209-19.1762-0.221919.6153S2-3.9560-18.6698-0.232519.0858μtr(s0-s1)-0.0061-0.0310-0.0806μtr(s0-s2)-0.52820.3345-0.0137VDA(s0-s1)/meV4.5VDA(s0-s2) /meV51
3.7 电荷转移和重组速率
将得到的Gibbs自由能、重组能以及转移积分分别代入方程(1),计算得到的电荷转移和电荷重组速率分别为:kCT= 7.2×1011s-1;kCR= 9.8×1010s-1.与实验中测得的速率范围非常吻合.
4 结 论
本文以实验报道的DR3TBDT/PC60BM体系为模型,在研究孤立给受体分子和复合物的基态和激发态性质的基础上,进一步研究了界面的激子分离和电荷重组.Gibbs自由能通过Rehm-Well表达式求得,重组能分别通过Marcus理论的双势阱和双球棍模型求得,转移积分通过广义的Mulliken-Hush模型近似求得,在此基础上根据Marcus电荷转移方程得到了该体系的电荷转移速率和重组速率分别为kCT= 7.2×1011s-1;kCR= 9.8×1010s-1.
