随机森林在基因调控网络推断上的比较与应用*
2019-03-19张铭智尤东方何文静张汝阳胡志斌
张铭智 尤东方 何文静 张汝阳,2 陈 峰,3,4 胡志斌 赵 杨,6,7△
基因调控网络(gene regulatory network,GRN)是当前功能基因组学所研究的重要内容之一,作为一种描述基因间相互作用关系的方式,在推断复杂疾病的致病原理过程中发挥着重要的作用。通过对基因调控网络的分析,我们能够更加深入地了解各基因的生物学功能、理解基因间的调控机理并推断出未知基因的功能,这对疾病诊断、临床实践、药物研发等方面有指导性的意义[1-2]。近年来,随着高通量生物实验技术的快速发展和计算机技术的进步,使得从高维生物组学数据中推断复杂的基因调控网络成为可能。大量推断方法应运而生:基于信息论方法[3-5]、基于模型方法[6-8]、基于监督或非监督学习方法[9-10]等等。这些方法主要是从基因表达数据中获取功能关系信息,用以模拟真实的网络结构,从局部甚至是整个基因组的意义上揭示基因间可能存在的相互关系[11]。
随机森林最早是由Leo Breiman和Adele Cutler共同提出的一种集成算法[12]。该算法不但能够很好地处理组学数据中存在的数据不完整、受噪声影响、高维低样本和基因间的非线性调控等问题,而且能够大幅度缩短运算时间。因此,越来越多的学者倾向于使用随机森林算法进行网络构建。如Huynh-Thu等[13]、Marit Ackermann等[14]均证明了随机森林的有效性和易扩展性。dynGENIE3(dynamical GENIE3)[15]和iRafNet(integrative random forest for gene regulatory network inference)[16]是两种具有代表性的扩展方法,在已有文献中未见两种方法和经典随机森林三者之间的比较,故本文旨在比较上述三种算法的网络重建能力,从而为提高推断性能提供参考性建议。
方 法
1.基于静态数据的随机森林算法
经典随机森林主要是从静态基因表达数据中推断调控网络。我们令X={X1,X2,…,XN}代表稳定状态下的基因表达值,其中Xk∈P,k=1,2,…,N代表p个基因在第k个状态下的表达值:假设每个基因xkj的表达受其他所有基因的共同调控,因此第j个基因的表达值可由如下函数表示:
(1)
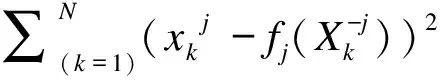
(1)采用Bootstrap方法从训练样本Xj={(xkj,Xk-j),k=1,2,…,N}中有放回地随机抽取N条观测得到一个子样本集合;重复上述抽样方法B次,得到B个子样本用于构建B棵独立的回归决策树。

(3)将所有的树进行汇总,根据树分裂时节点方差平均减少值(IncNodePurity)的大小来判断特征对基因j的影响程度:该值越大,说明对应的特征对基因j存在调控的可能性越大。
(4)按步骤(2)和(3)分别计算p个基因与待选特征之间的节点方差平均减少值(IncNodePurity),将p(p-1)个节点方差平均减少值从大到小排序:排名越靠前,则越有把握认为真实网络中存在对应的调控关系。最后通过与金标准的比较来评价算法的性能。
2.基于动态数据的随机森林算法
动态数据包含了时间历程,显示了网络在受到外界干扰时做出的反应以及干扰移除后回归稳态水平的动态变化过程,理论上更能说明基因调控的方向性和因果关系。因此有学者提出了一种利用时间序列数据进行网络推断的方法dynGENIE3。该算法与经典随机森林算法的区别在于训练样本的构造不同:令DTS={X(t1),X(t2),…,X(tN)}代表时间序列数据中各时间点的表达值(TS:time-series),其中X(tk)∈p,k=1,2,…,N代表p个基因在第k个时间节点的表达值:X(tk)=(X(tk)1,X(tk)2,…,X(tk)p)T。假设基因j在tk时刻的变化率受当前时刻所有基因的影响,即满足常微分方程:
(2)
动力学参数αj代表基因j的衰减率(decay rate)。当时间序列数据中各个相邻时间点的间隔一致时:
αj=1/(t(k+1)-tk)
(3)
通过(2)和(3)可得到基因j在t(k+1)时刻表达值的估计:
(4)
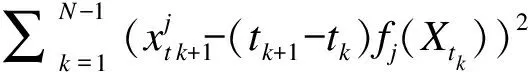
(5)
3.结合静态数据和动态数据的随机森林算法
静态数据和动态数据是从不同角度体现网络的内在结构,同时利用这两种数据进行网络推断能够更加全面地反映出真实网络状态。
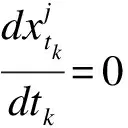
(6)
此时训练样本可表示为:
(7)
将该训练样本与算法2中的训练样本合并:
(8)
基于LSj进行网络推断即可同时利用两种数据中蕴含的基因关联信息。

(9)
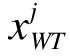
(10)
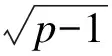
4.算法的评价标准
将得到的结果与金标准进行比较,把推断结果分为四类:真阳性(true positive,TP)、假阳性(false positive,FP)、真阴性(true negative,TN)以及假阴性(false negative,FN),如表1所示。则特异度(specificity,Sp)、灵敏度(sensitivity,Se)、查全率(recall,R)和查准率(precision,P)的计算公式如下:
(11)
(12)
(13)

(14)
本文主要通过受试者工作特征曲线线下面积(Area under ROC,AUC)以及精度召回率曲线线下面积(Area under PR,AUPR)的大小来衡量不同算法的优劣性。
以上所有算法的网络构建及性能评价均由R-3.5.1软件实现:经典随机森林使用‘randomForest’包构建;dynGENIE3[15]和iRafNet[16]算法分别使用对应文献提供的R包构建;AUC和AUPR通过‘ROCR’包计算所得。
数据来源
本文模拟数据来自Dialogue for Reverse Engineering Assessments and Methods第四次竞赛(DREAM4),包含5个10基因规模和5个100基因规模的网络调控数据,每个网络均含有静态数据和动态数据,且所有数据都经过归一化处理。其中静态数据包括野生型数据(wild type)、基因敲除数据(knock out)和基因敲低数据(knock down);野生型数据为稳定状态下各基因的表达量、敲除数据是抑制某基因时其余基因的表达情况、敲低数据则是在某个基因表达强度减半时其余基因的表达情况。动态数据为时间序列数据(time series),反映了稳态网络在受到外界干扰及干扰去除后所有基因表达量的变化情况,测量的时间间隔为50秒,共测量21次。同时还提供了这10个网络中真实存在的调控关系(金标准),用于评价算法的优劣性。所有的模拟数据和金标准可在DREAM4官网上下载(http://dreamchallenges.org/project-list/dream4-2009/)。验证数据为DREAM5提供的大肠杆菌基因表达微阵列数据,包括805个微阵列芯片,4511个基因的表达值。基因表达数据及每个微阵列芯片的详细描述均可在DREAM5官网中获取(https://www.synapse.org/#!Synapse:syn2787209/wiki/70349)。
结果与分析
从DREAM4参赛队伍的结果中发现,使用基因敲除数据推断网络的结果普遍优于从其他静态数据中推断的结果,这可能是因为基因敲除数据中蕴含的信息更加丰富[17]。因此,对于静态数据我们主要从基因敲除数据来推断网络。
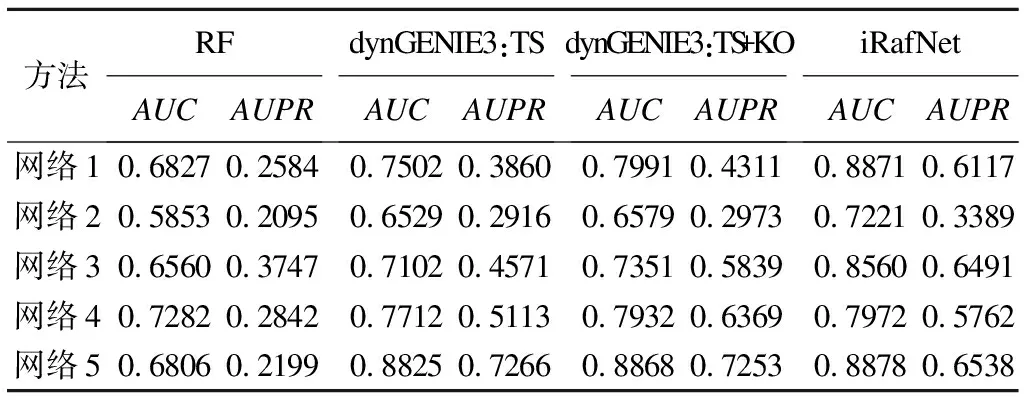
表2 比较各种方法在10基因规模网络中的AUC和AUPR值
TS:时间序列数据;KO:基因敲除数据

表3 比较各种方法在100基因规模网络中的AUC和AUPR值
TS:时间序列数据;KO:基因敲除数据
由表2可知:当网络结构较为简单时,dynGENIE3和iRafNet算法在5个网络中的ROC和PR曲线线下面积相比于随机森林均得到了明显的提升。同时发现dynGENIE3:TS和dynGENIE3:TS+KO曲线下面积基本一致,说明在该情况下,基因敲除数据相较于时间序列数据包含较少的额外信息。
由表3可知:当网络结构变复杂时,dynGENIE3和iRafNet算法在各个网络中都能提高PR曲线下面积。比较dynGENIE3:TS和dynGENIE3:TS+KO可发现当结合基因敲除数据进行网络推断时ROC曲线下面积明显增加:5个网络分别增加了19.40%、29.59%、27.13%、26.97%和15.66%(以ROC曲线下面积等于0.5为基线),且后者的PR曲线下面积也分别增加了0.0579、0.0388、0.0696、0.0523和0.0286。进一步比较dynGENIE3:TS+KO和iRafNet:对于ROC曲线下面积除了网络5,iRafNet均优于dynGENIE3:TS+KO;对于PR曲线下面积iRafNet均明显大于dynGENIE3:TS+KO。
为了进一步验证以上结果,我们分别用这三种方法对大肠杆菌的基因表达数据构建网络并计算AUC和AUPR值。结果如下:RF对应的AUC和AUPR值分别为0.5633和0.030;dynGENIE3:TS为0.5579和0.019;dynGENIE3:TS+KO为0.5837和0.031;iRafNet为0.6482和0.102。无论是AUC还是AUPR值,iRafNet算法均优于dynGENIE3和经典随机森林算法;并且结合基因敲除数据的dynGENIE3算法能够有效提高网络预测的准确性。与模拟数据的结果基本一致。
讨 论
基因调控网络的构建已经成为当前研究的热点领域之一,是对不同组学数据进行处理和挖掘的过程,将表达数据转换成由若干节点和边组成的图形,为进一步深入了解节点间复杂的调控关系和作用机理提供了参考模型。本文基于模拟数据和大肠杆菌基因表达数据对经典随机森林(RF)、dynGENIE3和iRafNet三种方法的网络推断能力进行了比较,这三种方法均将含有p个基因的网络推断问题转化为p个不同的特征选择问题。从100基因大小网络的推断中我们发现:RF算法对应的ROC曲线下面积大于dynGENIE3:TS,与前文提到的利用动态数据推断网络更加有效这一说法相矛盾。主要原因是时间序列数据中只包含21个时间点的表达值,不能够充分地反映各基因间存在的调控关系。而dynGENIE3:TS对应的PR曲线下面积更大,说明其推断结果的覆盖率和准确率比经典随机森林更高。故当时间序列数据提供更多时间点的基因表达测量值时,理论上其推断性能将优于静态数据。对于dynGENIE3算法,在结合静态数据后ROC和PR曲线下面积均得到了提升,这表明同时结合多种数据进行网络推断确实能够提高算法的性能。虽然iRafNet和dynGENIE3:TS+KO都是结合动静态数据网络推断算法,但两者的性能却存在着差异:dynGENIE3直接将基因敲除数据作为训练样本的一部分,每棵树都利用部分信息;而RafNet则是将基因敲除数据转换成抽样权重,使得每次分裂时都能充分利用数据中蕴含的信息。因此,如何充分提取数据中的信息将是提高算法性能的另一重要途径。
综上所述,随机森林是一种高效便捷的机器学习方法,不但能识别出基因间存在的非线性关系,也能同时整合多种不同类型的数据,在未来的研究中还可结合单核苷酸多态性(single nucleotide polymorphisms,SNP)、甲基化表达等数据进一步完善基因调控网络。随机森林本身也存在着不足,该算法无法很好地分离出网络中的直接调控效应和间接调控效应:当真实网络中存在i→k→j通路时,很可能会错误地认为基因j是受基因i直接调控的:i→j,使推断结果中假阳性比率上升,整体精确率下降;而在i→k→j这一通路的基础上如果同时存在i→j,又会降低i→j、k→j的推断效能,使查全率下降[18],共同导致PR曲线下面积的减少。另一方面,如果基因i同时对基因j和k有着调控关系,即j←i→k,将会导致基因j和k之间产生虚假关联:j←k或j→k。因此,如何将直接效应和间接效应分离,如何对算法产生的结果进行假设检验并剔除虚假关联和混杂将是提高算法性能的重要研究方向。
