金属离子水热法掺杂δ-MnO2及其催化氧化氯苯性能
2019-03-13罗邯予胡立扬王恺莹陈远庭张婷婷
罗邯予 胡立扬 王恺莹 陈远庭 张婷婷
(北京化工大学化学工程学院,北京 100029)
0 引 言
冶金、化工和垃圾焚烧等工业过程排放的废气中含有大量的氯代有机污染物,如氯苯、二噁英、多氯联苯等,这些含氯挥发性有机物(CVOCs)具有持久性,可破坏臭氧层,是雾霾和光化学烟雾生成的诱因之一,并且可在人体内长时间停留积累,严重威胁人类身体健康[1-2]。联合国环境规划署发布的斯德哥尔摩公约中有12种含氯有机化合物被列为持久性有机污染物[3]。因此,含氯挥发性有机物的控制排放已经引起各界的广泛关注,废气中CVOCs的降解技术已成为环境污染治理的焦点问题之一。
催化燃烧可以将难降解的CVOCs选择性氧化为CO2、HCl和H2O,具有催化效率高、节能环保、无二次污染等特点[4],是目前国内外最广泛使用的技术之一。催化燃烧技术的核心是催化剂的选择,要求所选用的催化剂具有价格低廉、催化效率高、热稳定性好等特点,处理CVOCs还须具有高的抗氯中毒能力。Mn作为可媲美贵金属催化活性的过渡金属[5-6],是应用最广泛的CVOCs催化燃烧处理催化剂,其中δ-MnO2因其特殊的层状结构和较大的比表面积,广泛应用于工业合成和污染治理领域。催化剂中掺杂少量其他元素,可以改变催化剂的物理化学性质,研究表明,MnO2中掺杂少量其他金属(Cu2+,Fe3+,Al3+,Co2+),可以制备出不同晶型与形貌的材料,改变MnO2的物理化学性质,提升催化剂的催化活性和抗中毒能力[7-10]。然而文献中将金属离子掺杂δ-MnO2应用到CVOCs的研究较少,尤其是掺杂金属离子价态和半径对MnO2晶型和形貌的影响缺少系统的研究。因此本文系统研究了不同金属离子掺杂δ-MnO2得到的催化剂的物理化学性质,并研究其催化燃烧降解CVOCs的性能及其作用机理。
1 实验部分
1.1 实验材料
无水乙醇、硝酸、高锰酸钾、九水合硝酸铝、硝酸铁(分析纯,北京化工厂);六水合硝酸钴(分析纯,西陇化工);硝酸铈(分析纯,天津津科精细化工);柠檬酸(分析纯,阿拉丁)。
1.2 催化材料制备
采用水热法制备金属离子掺杂层状锰。按照金属离子与锰离子的物质的量之比1∶2,分别称取一定量硝酸铜、硝酸铝、硝酸钴、硝酸铁和硝酸铈加入到一定量浓度为0.01 mol·L-1的高锰酸钾溶液中,混合均匀后再加入2 mL 0.01 mol·L-1的柠檬酸溶液,均匀混合后,依次倒入聚四氟乙烯水热反应釜,温度控制在160℃反应12 h。将釜中得到的产物用去离子水洗涤3次后再用无水乙醇洗涤3次,再经12 h 80℃烘干,得到一系列不同金属离子掺杂δ-MnO2的催化剂, 分别记为 CuMn、AlMn、CoMn、FeMn、CeMn。 δ-MnO2的制备方法同上,无需添加金属离子。
1.3 催化材料表征
X射线衍射仪(XRD)采用Rigaku UltimaⅣ型,管电压 40 kV,管电流 30 mA,Cu靶 Kα射线(λ=0.154 06 nm),扫描速度 5°·min-1,扫描范围 5°~60°,日本Rigaku公司生产;比表面和孔隙度分析仪(BET)使用Quantachrome NOVA2000e型,美国康塔仪器公司生产,样品在真空条件120℃下处理2 h,液氮温度(-196℃),吸附质为高纯氮;扫描电子显微镜(SEM),HITACHI S-4700,加速电压 0.5~30 kV,二次电子检测器;X射线光电子能谱仪(XPS)使用Thermo ESCALAB 250型,Al靶Kα射线,束斑为150 W。透射电子显微镜(TEM)使用JEOL JEM-3010型,工作电压300 kV,样品在无水乙醇中超声30 min分散均匀;氢气升温程序还原 (H2-TPR)采用PCA-1200型,北京彼奥德电子技术有限公司生产,称取100 mg催化剂于化学吸附仪的石英管中,150℃氩气氛围预处理1 h,随后氩气吹扫冷却至室温,切换H2体积分数为5%的氩氢混合气体,气体流量30 mL·min-1,按照 10℃·min-1升温程序升至 800℃,热导检测器检测。
1.4 催化材料性能测试
催化剂的催化燃烧活性评价在固定床反应器中进行[11]。选用氯苯作为含氯挥发性有机物的模型化合物,催化剂添加量为0.2 g,空速为15 000 h-1,模拟富氧环境时,20%(V/V)O2,催化燃烧反应温度梯度范围为240~350℃,出气经六通阀自动进样至气相色谱进行检测,型号为GC2010plus:日本SHIMADZU 公司,色谱柱:RTX-5,美国 Agilent公司生产,进样口温度:200℃;升温程序:90℃保温1 min,以10℃·min-1的速度升温至120℃,并保温3 min,检测器温度:200℃,氢离子火焰检测器。氯苯去除率用η计算。

其中 C0为氯苯初始浓度,mg·m-3;Ct为催化反应温度为 t时氯苯出口浓度,mg·m-3。
2 结果与讨论
2.1 催化剂活性测试
图1给出了不同金属离子掺杂δ-MnO2得到催化剂的氯苯催化燃烧性能。结果显示,在相同反应条件下对氯苯的催化效率由大到小为:AlMn>FeMn>CeMn>CuMn>δ-MnO2>CoMn。
从图 1可以看出,Al3+和 Fe3+掺杂 δ-MnO2制得催化剂AlMn和FeMn氧化氯苯的效果最好,Cu2+和Co2+掺杂δ-MnO2制得催化剂CuMn和CoMn氧化氯苯的效果较差,Ce4+掺杂δ-MnO2制得催化剂CeMn在240~300℃氧化氯苯的效果较好。Fe3+改性后制得催化剂的低温活性大大提升,在240℃时氯苯的去除率可达98.7%,比单纯δ-MnO2对氯苯的去除性能提高了20.0%;Al3+和Ce4+改性后制得催化剂在240℃对氯苯的去除性能分别提高了14.2%和13.3%。催化材料FeMn虽然具有较高的氧化还原能力和低温催化活性,能高效降解氯苯,但是催化燃烧的温度对催化材料的性能影响不大,可能是由于FeMn在高温下容易结焦积碳失活。下文将从金属离子掺杂前后催化剂的物理化学性质出发,对金属离子掺杂得到催化剂的性质与活性之间的联系进行深入探讨。

图1 不同离子掺杂改性δ-MnO2得到催化剂的氯苯催化燃烧活性Fig.1 Catalytic performance of δ-MnO2catalysts obtained with different doping ions
2.2 金属离子改性δ-MnO2制得的催化剂表征
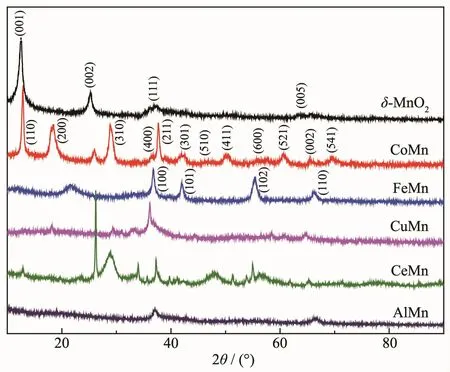
图2 不同离子掺杂改性δ-MnO2得到催化剂的XRD图Fig.2 XRD patterns of δ-MnO2catalysts obtained with different doping ions
图2 为催化剂 CuMn、AlMn、CoMn、FeMn、CeMn、δ-MnO2的XRD图。文献表明,采用相同的水热法时,若不加入金属离子进行改性,制备出的是δ-MnO2,且掺杂金属离子的离子半径和离子价态会改变催化剂的晶型[9-10]。由图2可知,当水热反应液中加入 Co2+后,(110)、(200)、(310)、(211) 等晶面特征峰尖锐,结晶度高,但是并未检测到氧化钴的晶相结构。研究发现,Co2+可以进入到锰氧化物的结构空缺位[12]。表1表明Co2+离子半径与Mn3+接近,进一步说明Co2+可能进入到δ-MnO2的锰氧八面体[MO6]内部[13],但(110)晶面所对应的2倍衍射角发生偏移,可能是由于Co2+离子的加入,导致δ-MnO2的锰氧八面体晶型结构轻微扭曲。当水热反应液中加入Fe3+后,(100)、(101)、(102)、(110)等晶面特征峰尖锐,说明催化剂结晶度高,但是并未检测到氧化铁的特征峰,说明Fe3+离子进入到锰氧化物的结构空缺位,能进入到层状锰的空缺位,可能是由于离子大小所决定,而层状锰晶型的变化,则可能是掺杂离子的价态特性及自身性质有关[14]。

表1 掺杂金属离子半径Table 1 Radius of doped metal ions
当水热反应液中加入Ce3+离子后,在催化剂CeMn的XRD图中检测到CeO2和α-MnO2的特征峰,在 CeO2(111)或者 α-MnO2(310)晶格面,对应特征峰 2θ=29.0°处,发生轻微的变化,同 Chen[15]所制得的催化材料共沉淀的3MnOx-1CeOy一样,反映了CeOx和MnOx的强烈相互作用,说明Ce4+并未进入锰氧化物的结构空缺位,而是在表面形成颗粒[16-17]。Ce4+不能进入锰氧化物的结构空缺位,可能是由于Ce4+离子半径较大。而当水热反应液中加入Al3+、Cu2+后,由XRD图可以看出,并未检测到δ-MnO2(001)、(002)、(111)和(005)晶面特征峰,说明 CuMn、AlMn并未形成MnO2锰氧化物典型晶型,这是由于Al3+、Cu2+离子半径较小,大量进入层间,导致层间作用力失衡而脱层[18-19]。掺杂的离子会因与锰离子的离子大小差异而进入不同的掺杂位点,由表1及XRD的晶相结构分析,可以推测得到不同金属掺杂进入不同位置的示意图,如图3所示。
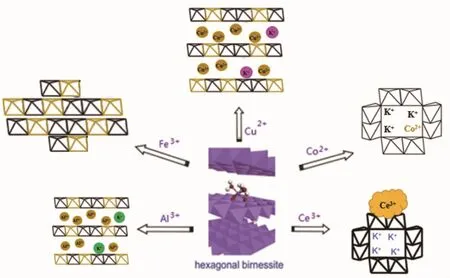
图3 金属离子掺杂入δ-MnO2位置示意图Fig.3 Schematic diagram of metal ions doping into δ-MnO2
2.3 催化材料组织结构分析
图4(a)为 催 化 剂 CuMn、AlMn、CoMn、FeMn、CeMn、δ-MnO2的N2吸附-脱附等温线。由图可知AlMn的孔容(Vpore)最大,在相对压力为0.5时,回滞环便出现,说明AlMn具有中低孔结构,其他各个催化剂的回滞环斜率 Q 大小为:QCuMn>QFeMn>QCeMn>Qδ-MnO2>QCoMn,回滞环斜率越大,说明催化剂孔容越大[20],能储存更多的气体,而回滞环随相对压力增大而吸附量越大,即吸附曲线斜率越大。催化燃烧降解烟气中含量较低的VOCs时,催化剂的孔隙不仅能吸附富集目标污染物,还可以吸附分子氧储存为Oads,在催化燃烧过程,能使更多的Oads转化为氧化活性位点[21],提高催化效率。所以,理论上回滞环越大,催化剂催化效果越优异。

图4 不同离子掺杂改性δ-MnO2得到催化剂的(a)N2吸附-脱附等温线和(b)孔径分布图Fig.4 (a)N2adsorption-desorption isothermal curves and(b)pore size distribution of δ-MnO2 catalysts obtained with different doping ions
表2列出了催化剂的比表面积与孔结构的数据,可以看出,催化剂AlMn的比表面积(SBET)最大(540 m2·g-1),是 δ-MnO2的 5.4 倍,而催化剂 CoMn 的比表面积最小。金属氧化物催化剂的比表面积一般小于200 m2·g-1[22],本研究制备的催化剂AlMn近似于活性炭的比表面积丰富度,这可能有助于提高催化活性;图4(b)为孔径分布图,由图4和表2可以看出,催化剂AlMn的孔径(D)主要分布在2~6 nm,催化剂CoMn的孔径主要分布在38~65 nm,各催化剂的孔径大小为:DAlMn≈Dδ-MnO2<DFeMn<DCeMn<DCuMn<DCoMn,催化剂内部孔道结构越致密,催化剂的活性位点越多,催化活性越佳。

表2 不同金属离子掺杂的δ-MnO2的比表面积、孔体积和孔径Table 2 Specific surface area,pore volume and pore diameter of δ-MnO2catalysts obtained with different doping ions
2.4 催化材料形貌分析
图5(a~e)是催化剂 CuMn、AlMn、CoMn、FeMn、CeMn的SEM图。图5(a)表明CuMn为丝状结构无序堆积而成。掺杂金属离子Al3+、Fe3+后得到的催化剂AlMn、FeMn的形貌均为纳米花球型结构,微球大小约为700 nm。这可能是由于Al3+、Fe3+均为+3价离子,与Mn3+、Mn4+价态更为相近,且离子大小也极为近似, 所以离子 Al3+、Fe3+能伪装成 Mn3+、Mn4+离子,进入二氧化锰六面晶体结构,并且不会造成催化剂表面形貌发生变化,仍然保持为纳米花球型。TEM表征(图6)可以看出,二者的花球内部差异较大,催化剂AlMn花球由片层结构堆积而成 “绣球型”,催化剂 AlMn(图(b1,b2))花球由絮状物质缠绕而成“棉花糖型”,而催化剂 FeMn(图(d1,d2))花球则是由颗粒状物质堆积而成“米花糖型”,这说明由于离子种类的不同,Al3+、Fe3+仍无法完全代替Mn3+、Mn4+离子形成晶格扭曲的二氧化锰六面晶体结构,所以造成这种外貌相似但本质不同的 “纳米花球型”材料。再结合比表面积可知“棉花糖型”的催化剂AlMn比表面积更大。催化剂CoMn的形貌为无序排列的柱状结构,柱子直径约为100 nm,但柱子表面有脱落片层和颗粒,且柱子大小不一,成型不佳。Co2+为+2 价,与 Mn3+、Mn4+的价态差异,会导致二氧化锰形貌发生改变。催化剂CeMn则是由无定形的颗粒和柱状结构组成。Ce3+也是+3价离子,但是离子半径较大,无法成功跻身进入二氧化锰六面晶体结构内部,所以造成催化剂形貌无法维持纳米花球型。由此可见,不同的金属离子掺杂的催化剂形貌有较大的变化。纳米花球型结构使AlMn和FeMn催化活性较高,而掺杂Co2+后成型不佳,使其催化活性低于改性前的δ-MnO2。

图5 不同离子掺杂改性δ-MnO2得到催化剂的SEM图Fig.5 SEM images of catalysts obtained with different doping ions

图6 催化剂的TEM图Fig.6 TEM images of catalysts
2.5 催化材料氧化还原能力分析
H2-TPR可以预判物质的氧化还原能力,对研究物质的催化氧化能力分析具有强有力的支撑。层状δ-MnO2的氢气还原峰在340~370℃左右,可通过掺杂金属离子后氢气还原峰的变化,推测其催化能力变化。

图7 不同离子掺杂改性δ-MnO2得到催化剂的H2-TPR图Fig.7 H2-TPR diagrams of δ-MnO2catalysts obtained with different doping ions
图7 为各金属掺杂层状锰后所得催化剂的H2-TPR图,催化剂的还原性能由大到小为:CuMn>AlMn≈FeMn≈CoMn>δ-MnO2≈CeMn。 不同金属掺杂进入δ-MnO2为催化剂的氢气还原峰的出现位置带来一定变化,金属Al3+掺杂层状锰后其还原峰出现在310和414℃,前者归属于催化剂的MnO2→Mn2O3还原峰,而后者应归属于Mn2O3→MnO还原峰[23];金属Fe3+掺杂层状锰后其还原峰出现在316、350和505℃,前2个峰应归属MnO2→Mn2O3还原峰及Mn2O3→MnO还原峰,而后者应归属于Fe2O3→FeO还原峰,说明掺杂金属铁离子后,锰的氧化还原能力显著增强,且该TPR剖面中观察到的另一个重要特征是在尖晶石结构中引入了3种还原峰,降低了其温度。这表明Mn的存在有利于铁素体的还原,提高了Fe3+和Mn阳离子对催化剂的氧化能力,这可能归因于Fe3+和Mn阳离子之间的强相互作用[24];金属Cu2+掺杂层状锰后其还原峰出现在285℃,归属于MnO2的还原,可以推测出CuO→Cu2O和Cu2O→CuO氧化还原转化能力极强,对比掺杂改性前,峰位产生温度变低了55℃,明显提前,其可能原因是Cu的掺杂与Mn形成了铜锰尖晶石结构,由与XRD结果一致[25];金属Ce4+掺杂层状锰后其还原峰出现在334和422℃,前者应归属于MnO2的还原,而后者应归属于CeO2的还原[26]。
2.6 催化材料活性物种分析
图8分别为催化剂 δ-MnO2、AlMn和 FeMn和催化降解氯苯前后的Mn2p、O1s的XPS图谱,遵循解卷积一般规律,经过分峰拟合处理得到催化剂催化降解氯苯前后表面活性组份的峰面积列于表3和表4。
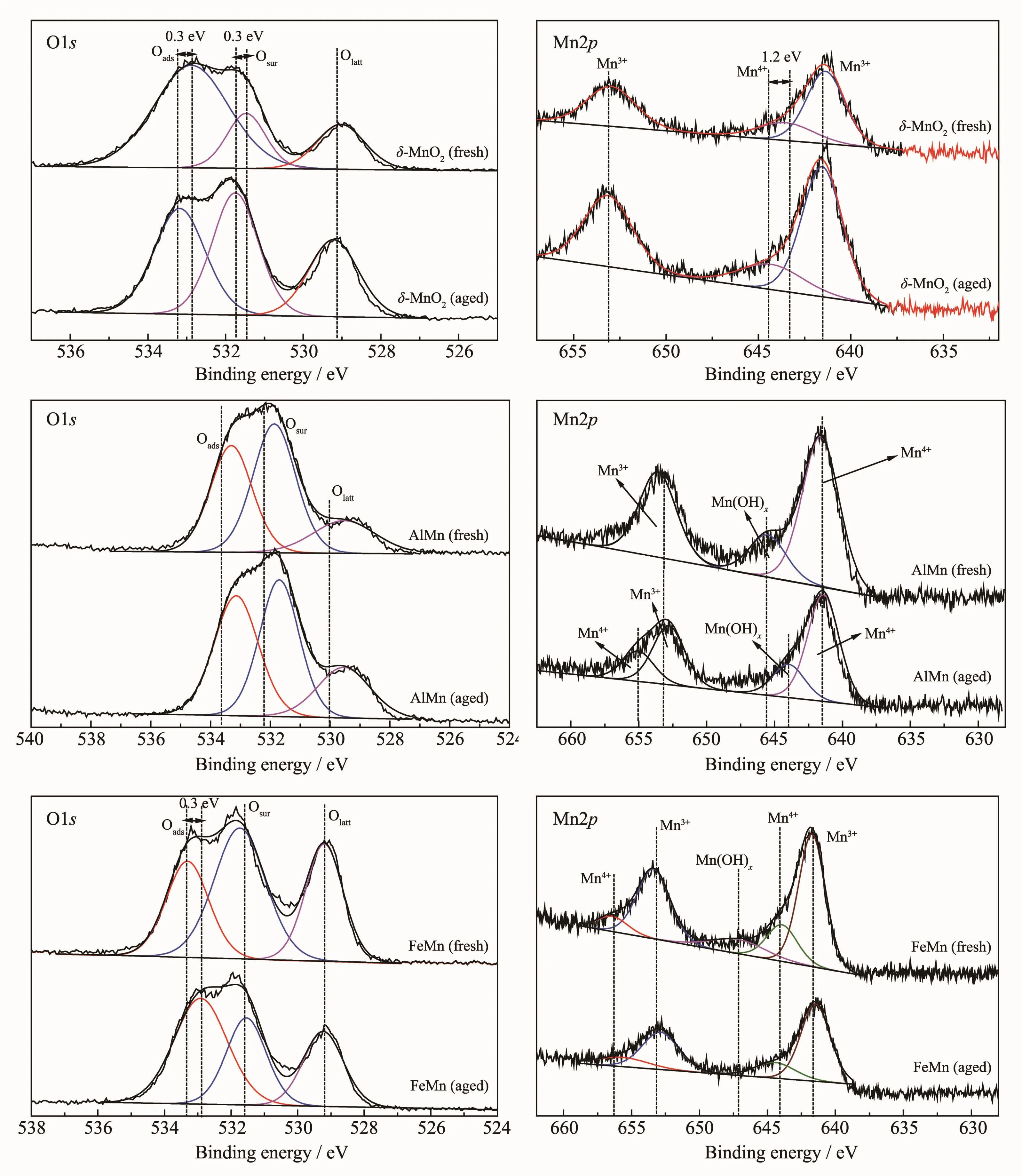
图8 新鲜与旧催化剂的活性物种的XPS对比谱图Fig.8 Contrast XPS spectra of active components for fresh and aged catalysts

表3 XPS表征得到的催化剂反应前后表面活性组份Mn2p的结合能位置及元素分析Table 3 Binding energy position and relative ratio of catalysis(fresh and aged)surface active components Mn2p obtained from XPS characterization
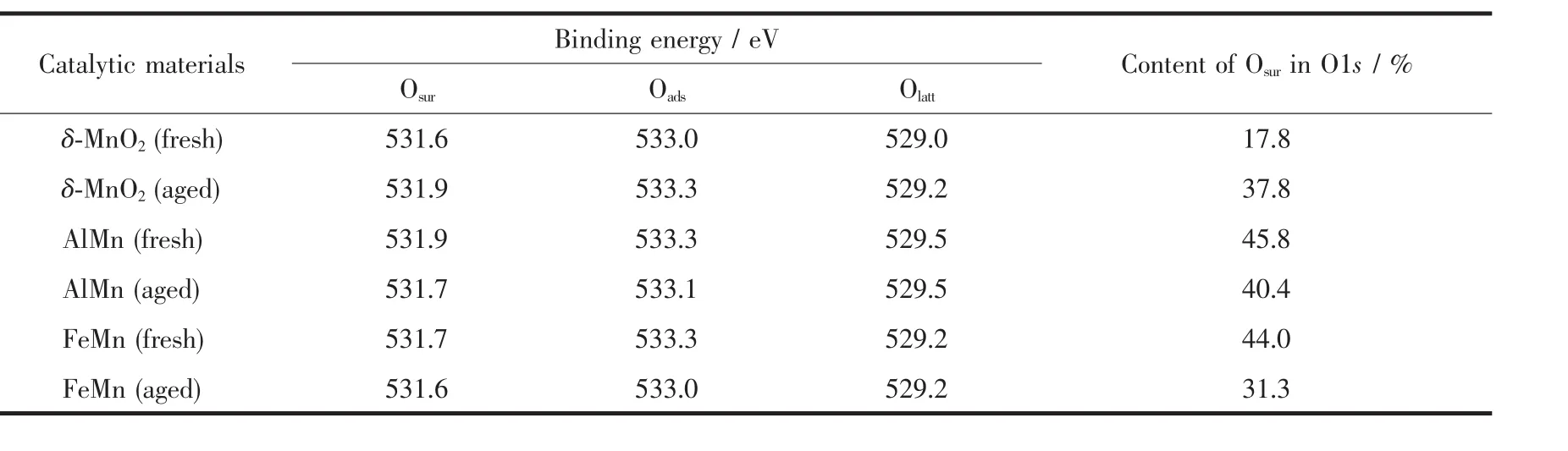
表4 XPS表征得到的催化反应前后表面活性组份O1s的结合能位置及元素分析Table 4 Binding energy position and relative ratio of catalysis(fresh and aged)surface active components O1s obtained from XPS characterization
由氯苯催化性能实验可知,相同反应条件下,对氯苯的催化效率为:AlMn>FeMn>δ-MnO2,结合表3可知,3种催化剂催化降解氯苯之后,Mn3+峰面积皆增大,说明催化降解氯苯过程中Mn4+转变为Mn3+,而3种催化剂中的含氧类活性物种丰富,其中新鲜 δ-MnO2含有 17.8%的 Osur、64.2%的 Oads、17.9%的 Olatt, 新鲜 FeMn含有 44.0%的 Osur、27.0%的 Oads、28.9%的 Olatt,新鲜 AlMn含有 45.8%的 Osur、37.1%的Oads、17.1%的Olatt。催化剂AlMn具有较高的催化降解氯苯的效率可能是由于其含有丰富的Osur,因为Osur的氧化能力最佳[27-28];催化剂使用之后δ-MnO2的Osur、Olatt含量增加,Oads减少,说明 Oads起主要的活性位点作用;使用之后的FeMn催化剂Olatt含量增加,Oads含量变化不大,Osur减少,说明Osur起主要的活性位点作用;而使用之后的AlMn的Oads、Olatt含量增加,Osur减少,说明Osur起主要的活性位点作用。Osur的比例增加右利于氧物种从晶格氧向表面氧转移,有利于氧化还原反应的进行。同时,由图8可知,层状锰δ-MnO2使用之后,Mn4+的特征峰键能增加1.2 eV,Osur的特征峰键能增加0.3 eV,Oads的特征峰键能增加0.3 eV,其键能的增加使得Mn4+不易向Mn3+转化,活性氧氧化能力减弱。
由此可以推断,AlMn在较宽温度范围都具有较高催化活性的原因是该催化材料具有最高含量的Osur和较低的可还原温度。此外,AlMn较高的比表面积和孔容可能有效抑制催化剂的中毒。
3 结 论
利用水热法制备出系列不同金属离子掺杂的δ-MnO2催化剂,获得了不同形貌的催化剂,并考察了其对氯苯的催化燃烧活性和失活原因。实验发现,掺杂离子的不同会造成δ-MnO2晶型形貌的变化,这与掺杂金属离子的价态和离子大小有关。AlMn具有较高的比表面积和“棉花糖”型特殊纳米花球形貌。相同反应条件下,不同金属离子掺杂改性得到催化剂对氯苯的催化效率比较结果为:AlMn≈FeMn>CeMn>CuMn>δ-MnO2>CoMn。N2吸附-脱附结果表明AlMn内部孔道结构致密,使催化剂的活性位点增多,催化活性较好。H2-TPR结果表明AlMn氧化还原性能较为优异。通过对新鲜催化剂和旧催化剂进行XPS分析,AlMn在较宽温度范围都具有较高催化活性的可能原因是该催化材料具有最高含量的Osur。
