易错PCR技 术定向进化褐藻胶裂解酶Alg-2的分析
2019-03-11赵春梅
李 树,张 伟,赵春梅
(1.山东大学(威海)海洋学院,山东 威海 264209;2.江南大学生物工程学院,工业生物技术教育部重点实验室,江苏 无锡 214122)
近年来,随着海洋科学研究和海洋资源开发的不断深入,褐藻胶裂解酶作为一种重要的海洋生物资源开发用酶越来越引起人们的兴趣与重视。利用褐藻胶裂解酶作用于褐藻胶获得的褐藻寡糖(alginate oligosaccharides,AOS),因具有特殊的化学特性和生物特性,在农业、食品、药品、保健品、化妆品、冶金、化工等领域具有潜在、广泛的应用价值,并且酶法制备AOS具有环保、清洁、条件温和、效率高等优点,其取代物理化学法制备AOS已成为必然[1-5];巨量的海藻生物质因不含陆生植物难以降解的木质素,且纤维素含量较低,易于进行生物处理,在转化乙醇方面已有报道[6-7],相比于纤维质原料制造乙醇具有显著的优越性,而褐藻胶裂解酶在这一领域中将发挥难以取代的基础性作用;在海藻遗传工程的研究中,由于褐藻多糖黏度极高,很难将DNA、单细胞、原生质体和胞内物质分离,而褐藻胶裂解酶的应用则有效排除了这种障碍,因而其在藻类生理生化、遗传学、分子生物学等基础研究领域中发挥重要的作用,这些领域均属当今生命科学研究的前沿和热点[8];除此以外,也有报道褐藻胶裂解酶在医学上已经作为药品酶应用于临床,治疗由致病菌铜绿假单胞菌引起的肺炎[9]。所以,在当今开发海洋资源战略的大背景下,对褐藻胶裂解酶的研究与应用,必将成为开发新功能性物质、新材料、新能源的重要基础。
迄今为止,国内外学者对褐藻胶裂解酶的来源、分类、酶学性质、酶的结构、酶催化机理等方面作了大量的基础研究[9-16],已报道的裂解酶近200 种。从已有的报道看,虽然研究者采用的酶活力测定方法有多种,酶活力定义方式及单位也不一致,但明显不容忽视的一个事实是,褐藻胶裂解酶存在发酵水平低、酶活力不高等问题。目前国际市场上只有美国Sigma公司商业化的褐藻胶裂解酶产品A1603,酶活力大于10 000 U/g,但价格昂贵,仅以试剂形式出售。尽管已有20多种裂解酶的基因得到克隆和测序,并构建出多种重组裂解酶基因工程菌,其主要目的还是获得单一酶组分,进而研究其酶学性质和催化特性,即使经过高密度发酵的调控优化,其表达量还处于较低的水平,例如酶活力仅为117、196 U/mL(3,5-二硝基水杨酸(3,5-dinitrosalicylic acid,DNS)法)[17-18];本课题组筛选到的海洋细菌Bacillus mesophilus sp. nov和Tamlana sp. S12,其发酵上清液褐藻胶裂解酶活力分别为46、126 U/mL(DNS法)[19];中国海洋大学筛选到的菌株HZJ216发酵上清液酶活力为338 U/mL(紫外法)[20]。已有报道的这些褐藻胶裂解酶及其生产菌株,目前还未见到官方认可的工业化生产及应用。
目前,半理性设计的蛋白质定向进化技术已经被广泛用于基因的体外进化,在改善酶的催化特性方面产生重要作用,而易错聚合酶链式反应(polymerase chain reaction,PCR)就是采用最多的一种定向进化技术。易错PCR技术是指通过改变PCR条件,例如改变Mg2+或者Mn2+,使用低保真度的Taq酶等,在某种程度下使碱基随机错配而产生不同的突变体,然后构建突变体文库以便筛选出具有目的表型的突变株。一般情况下,经过1轮易错PCR很难筛选出具有优良性状的目的基因,往往将第1轮筛选获得的有益突变基因作为下一轮易错PCR扩增的模板,连续随机的突变,最终筛选出所需的有益突变,该技术已经在提升脂肪酶、木聚糖酶、谷氨酸脱羧酶、精氨酸脱亚胺酶等酶活力方面取得了成功,对研究酶三维结构中关键氨基酸位点也起到了重要的指导作用[21-22]。
本研究采用易错PCR技术对海洋细菌Tamlana sp. S12中具有代表性的褐藻胶裂解酶组分Alg-2进行2 轮体外定向进化,通过96 孔板高通量筛选分别获得褐藻胶裂解酶活力提高和降低的突变株,通过与母本进行碱基和氨基酸序列比对,发现与酶活力相关的氨基酸位点,不仅有助于深入理解褐藻胶裂解酶的催化机制,更为后续利用理性设计手段改造褐藻胶裂解酶提供理论参考。
1 材料与方法
1.1 材料与试剂
海洋细菌Tamlana sp. S12为本实验室保藏;褐藻胶裂解酶Alg-2(基因登录号ABD808071)、质粒Escherichia coli DH5α、Rosetta agami(DE3)、pET-30a(+) 宝生物工程(大连)有限公司;DNA提取和质粒提取试剂盒 北京博大泰克生物基因技术有限责任公司;Taq DNA聚合酶、限制性内切酶、T4 DNA连接酶 上海生工生物技术服务有限公司;PCR试剂盒 德国Qiagen公司;基因标准Marker 美国Thermo Scientific公司;牛血清蛋白、琼脂糖 美国Sigma公司;其他试剂均为国产分析纯。
1.2 仪器与设备
PCR仪 美国Bio-Rad公司;DYY-6B琼脂糖凝胶电泳仪、VC950型超声波细胞破碎仪 美国Sonics公司;AB204-N分析天平 瑞士梅特勒-托利多有限公司;3K15型高速冷冻离心机 美国Sigma公司;LC-15C高效液相色谱 日本岛津公司;CMax Plus酶标仪 日本Hitachi公司;AKTA蛋白纯化系统 美国GE公司;UV-2100紫外-可见分光光度计 美国Unico公司。
1.3 方法
1.3.1 易错PCR扩增
分别采用不同浓度的Mg2+(5、6、7、8、9、10 mmol/L)和Mn2+(0.02、0.05、0.1、0.15、0.2、0.25 mmol/L)对褐藻胶裂解酶Alg-2进行易错PCR扩增,并通过两者的不同浓度配比进行优化。根据Alg-2的基因序列设计引物为F:5’-TACTCAGGATCCTCTACTGATG ATGATTTAGTTGA-3’;R:5’-TACTCAAAGCTTCTAT AACGTGGTATGCGATT-3’。
PCR体系为50 µL(Mg2+和Mn2+除外):1×Taq DNA聚合酶缓冲液、0.2 mmol/L dATP、0.1 mmol/L dGTP、0.5 mmol/L dCTP和dTTP、引物30 pmol、模板 DNA 1 μg、Taq DNA聚合酶3 U。
PCR扩增条件:95 ℃预变性3 min;95 ℃变性55 s,56 ℃退火40 s,72 ℃延伸1 min。
1.3.2 突变体文库的构建
易错PCR的扩增产物电泳检测合格后,试剂盒切胶回收。用限制性内切酶BamHI和XhoI分别对载体pMDTM18-T和易错PCR条带进行消化,将Alg-2基因与线性化载体片段连接,16 ℃过夜,将连接产物电击法转入扩增宿主E. coli DH5α中,涂布含硫酸卡那霉素(50 mg/L)的抗性平板构建突变体文库。
1.3.3 突变文库的筛选
96 孔板中每孔装有1 mL灭菌LB培养基,对应一株转化子,并且将野生型Alg-2(亲本)作为对照。30 ℃、200 r/min培养12 h,所得菌液即为粗酶液。利用排枪快速移取100 μL粗酶液至96 孔PCR板中,注意每孔一一对应,孔中事先加入底物溶液100 μL(1%褐藻酸钠溶液),于45 ℃水浴30 min,再加入200 μL的DNS溶液100 ℃水浴5 min,显色,利用酶标仪在540 nm波长处测定吸光度。发酵上清液的粗蛋白含量采用考马斯亮蓝法测定[11-12]。酶的比活力单位(U/g)定义为:每分钟产生1 μmol还原糖(以葡萄糖计)所需的蛋白量。以亲本酶活力(2 675.2 U/g)为参考,将高于或者低于酶活力20%以上的转化子视为突变体,进行斜面保藏。
1.3.4 褐藻胶裂解酶动力学Km值测定
Km值的测定采用双倒数作图法[23]。
1.3.5 褐藻胶裂解酶三维结构模拟及突变位点分析
筛选获得的突变体与亲本进行基因测序后,根据碱基序列生成氨基酸序列,分析碱基及氨基酸的突变情况。在SWISS-MODEL数据库(http://swissmodel.expasy.org/workspace)中输入各氨基酸序列生成褐藻胶裂解酶Alg-2的三维结构,分析其位点突变效应。
1.3.6 定点突变验证
为验证相关氨基酸的突变效应,选取酶活力正突变株P2-81,委托华大基因(北京)针对Lys286进行定点突变;选取酶活力负突变株N2-47,针对Gly60和Asp275进行定点突变。以重组质粒E. coli DH5α为模板设计引物,F和R为基因上下游引物,Fm和Rm为突变位点的上下游引物。以Lys286的定点突变为例,引物F同1.3.1节,Fm:5’-GGATCCTTAATACTTCGAAGGACCTG-3’;Rm:5’-AGTGATCGGAGGTTAGTAAGCTAATG-3’;引物R同1.3.1节。其中下划线部分为SalI的酶切位点,由CGAAGG突变成CGGAGG。分别以F/Rm和Fm/R为引物,扩增出含有突变位点的片段,构建重组表达质粒并测序[6,24]。
1.4 数据分析
PCR电泳照片用Photoshop CS6处理,相关数据用软件Origin 8.0作图,基因碱基的突变位点采用DNAMAN 8.5进行对比,褐藻胶裂解酶的三维结构采用SWISSMODEL模拟,其他数据采用Microsoft Office Word 2010处理。
2 结果与分析
2.1 Mn2+和Mg2+对易错PCR的影响
易错PCR是通过改变常规的PCR条件,在某种程度下使碱基随机错配而产生不同的突变体,而调整体系中的Mg2+或者Mn2+浓度则是目前常用的方法[25]。分别采用5、6、7、8、9、10 mmol/L Mg2+对Alg-2进行PCR扩增,由图1可知,随着Mg2+浓度的升高,PCR电泳条带的亮度也逐渐增加,在Mg2+浓度为9 mmol/L和10 mmol/L时结果较为理想。
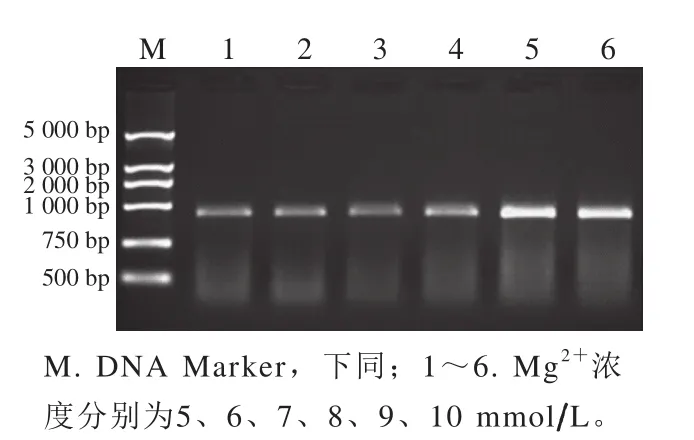
图1 Mg2+浓度对易错PCR结果的影响Fig. 1 Effect of Mg2+ concentration on error prone PCR
Mn2+可以通过降低聚合酶对模板的特异性而增加碱基错配概率。选取0.02、0.05、0.1、0.15、0.2、0.25 mmol/L不同浓度的Mn2+进行易错PCR,由图2可知,所选范围内的Mn2+浓度对PCR影响不大,电泳条带的亮度均比较理想。
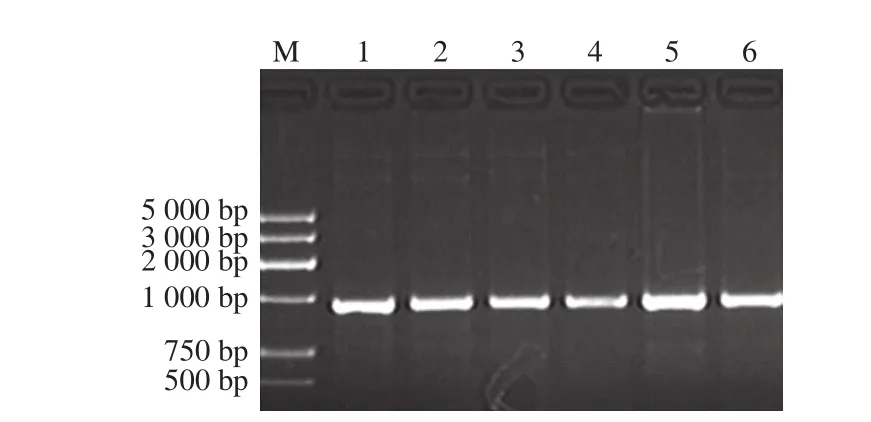
图2 Mn2+浓度对易错PCR结果的影响Fig. 2 Effect of Mn2+ concentration on error prone PCR
选择Mg2+(9 mmol/L和10 mmol/L)和Mn2+(0.05、0.1、0.15 mmol/L)的不同浓度组合,进行PCR构建突变文库,从每组突变文库中随机抽取5 株菌进行Alg-2的基因测序以计算突变率,如表1所示,其中梯度2(Mg2+9 mmol/L、Mn2+0.1 mmol/L)的平均突变率为0.27%,接近0.25%的范围,即每个基因序列有4 个碱基突变,这是目前公认的合适突变率,而其他梯度的突变率均偏低或者偏高,弃用。

表1 不同浓度Mg2+和Mn2+组合的PCR突变率Table 1 PCR mutation rates with different concentrations of Mg2+ and Mn2+
2.2 突变文库的筛选结果
以褐藻胶裂解酶Alg-2基因组为模板进行第1轮易错PCR。将重组质粒pMDTM18-T电转化转入扩增宿主E. coliDH5α中,筛选获得突变文库5 000 株菌。利用96 孔板发酵配合酶标仪测酶活力高通量筛选酶活力突变菌株。由表2可知,相比于母本Alg-2的酶比活力2 675.2 U/g,1株正向突变株P1-37(酶比活力4 332.5 U/g)和1株负向突变株N1-42(酶比活力1 417.4 U/g)被筛选获得。分别以P1-37和N1-42的基因组为模板再进行第2轮易错PCR,突变文库容量仍然为5 000 株菌。经过96 孔板发酵及酶活力测定,在以P1-37为模板的突变株中获得1株酶活力进一步提高的菌株P2-81,褐藻胶裂解酶比活力达到6 420.8 U/g,是母本的2.41 倍;而在以N1-42为模板的突变株中获得1株酶活力更低的菌株N2-47,其活力降低至294.2 U/g,只有母本的11.2%,上述结果证明易错PCR技术在改进酶活力方面确实存在极大的优势。
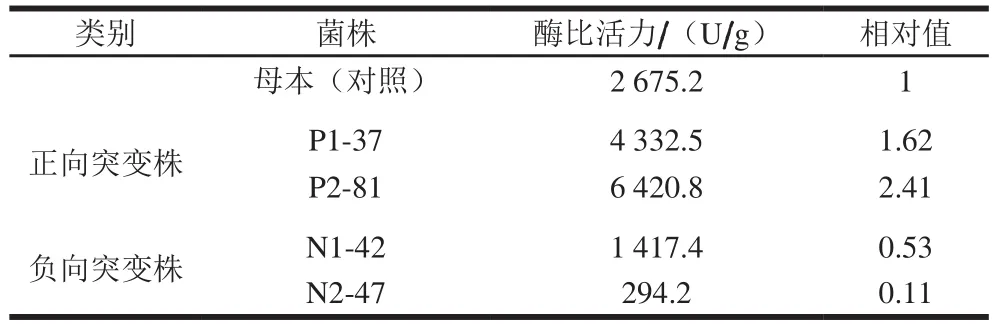
表2 两轮易错PCR筛选结果Table 2 Results of two screening rounds with error-prone PCR
2.3 突变菌株的基因测序结果
对2轮易错PCR筛选获得的4 株褐藻胶裂解酶Alg-2基因突变株进行基因序列分析,结果表明,P1-37中有3 个碱基发生突变,并且碱基的变化均引起氨基酸的改变(表3)。在P1-37发生正向突变的基础上进行第2轮易错PCR,筛得的P2-81又有3 个碱基发生突变,除A486G外,其余2 个突变均引起氨基酸的改变。而对于负突变株N1-42,则有4 个碱基突变,其中T417A的突变编码均为Arg,其余3 个都导致氨基酸的改变。在N1-42的基础上再次进行易错PCR,获得的N2-47中又有2 个碱基发生突变,其中的T285A为无义突变。上述正突变和负突变的测序结果为解析Alg-2褐藻胶裂解酶活力的变化提供依据。

表3 易错PCR突变菌株的基因测序结果Table 3 Gene sequencing of mutant strains with error-prone PCR
2.4 褐藻胶裂解酶的Km值
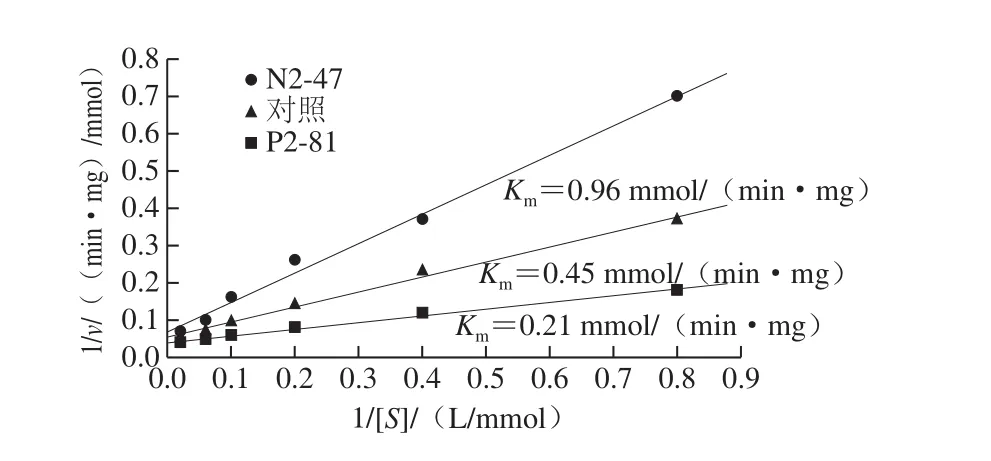
图3 Alg-2母本及正负突变株的Km值测定Fig. 3 Km values of Alg-2 of parental strain and positive and negative mutants
如图3所示,母本Alg-2的Km值为0.45 mmol/(min·mg),而酶活力提高的正突变株P2-81,其Km值下降为0.21 mmol/(min·mg),Km值下降表明褐藻胶裂解酶对底物的亲合力提高,即酶的催化效率提高。而负突变株N2-47,其Km值上升至0.96 mmol/(min·mg),表明酶对底物的亲和力下降,酶活力也相应地明显下降。
2.5 褐藻胶裂解酶的突变位点分析与结构模拟
将褐藻胶裂解酶Alg-2的亲本和突变株的氨基酸序列通过SWISS-MODEL REPOSITORY进行三级结构模拟。由图4可知,Alg-2富含β片层结构,属于典型的PL7家族。由于酶的结构具有较强的稳定性,少数氨基酸的突变不会引起三维结构的明显改变,所以Alg-2的亲本和突变株从三级结构上基本一致,仅存在细微方面的差别。在正向突变株P1-37中,43位点的Gly转变成Glu、188位点的Ile转换成Thr、250位点的Ile转换成Ser,这3 个位点均由极性较弱的氨基酸变成极性较强的氨基酸,亲水性增强,其与褐藻多糖的结合力增强,因而是酶活力提高的重要原因[26]。在P1-37的基础上获得的P2-81,60位点的Gly被Asp取代,氨基酸的解离常数增大,同样增加酶与底物的亲和性[27]。275位点的Asp虽然被Tyr取代,酸性减弱,但是Tyr作为质子的供体,是褐藻胶裂解酶完成β消除反应所必需的氨基酸[28],这可能是P2-81酶活力进一步增强的原因。对于负向突变株N1-42,有4 个碱基发生突变,但引起3 个氨基酸的改变,为Leu74Phe/Lys221Asn/Tyr308Cys。在此基础上进行第2轮易错PCR获得的N2-47,又有2 个碱基发生突变,但只引起了一个氨基酸的改变,为Lys286Glu。根据相关报道,褐藻胶裂解酶PL7家族中β片层结构域中有3 个保守区域,而Lys在保守区域中发挥着重要的作用[29-30]。由此推测2 株负向突变株中Lys被替换是导致Alg-2酶活力急剧下降的重要原因。
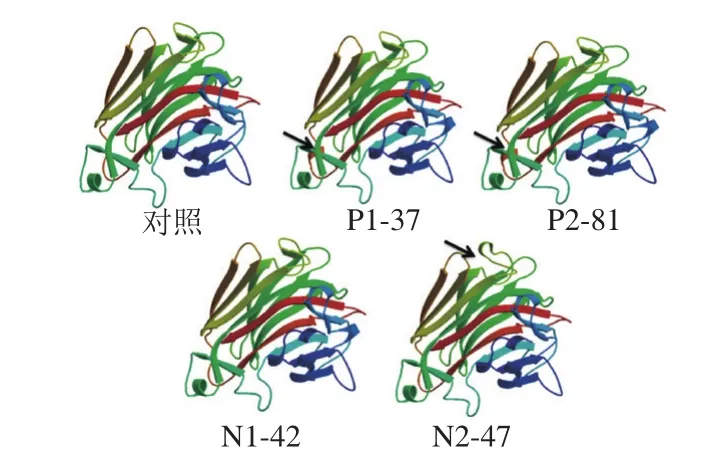
图4 褐藻胶裂解酶Alg-2三维结构模拟Fig. 4 Three-dimensional structure simulation of Alg-2
2.6 定点突变验证结果
为验证上述部分氨基酸位点的突变效应,选取酶活力正突变株P2-81,委托华大基因(北京)针对Lys286进行定点突变;选取酶活力负突变株N2-47,针对Gly60和Asp275进行定点突变。以Lys286定点突变为例,利用F/Rm和Fm/R为引物扩增出含有突变位点的片段,构建重组质粒并由华大基因进行测序,由图5可知,成功由CGAAGG突变成CGGAGG,即Lys突变成Glu。将重组质粒pMDTM18-T电转化转入扩增宿主E. coli DH5α中构建P2-81的突变株,发酵结果表明突变株的褐藻胶裂解酶的比活力为2 946.8 U/g,相比于母本下降幅度为54.1,这就进一步验证上述假设,即保守区域中Lys的被替换是导致Alg-2酶活力急剧下降的重要原因。对于负突变株N2-47,针对Gly60的定点突变尚在条件优化中,而对于Asp275的定点突变已经取得成功,突变株中Asp突变成Tyr,发酵检测其褐藻胶裂解酶的比活力由294.2 U/g提高到386.4 U/g,提高幅度为31.2%。这也进一步验证上述假设,Tyr作为质子的供体,是褐藻胶裂解酶完成β消除反应所必需的氨基酸[24],从而使酶活力增强。本实验室将继续采用更多轮次的易错PCR技术对褐藻胶裂解酶Alg-2进行定向进化并验证更多位点氨基酸的突变效应。
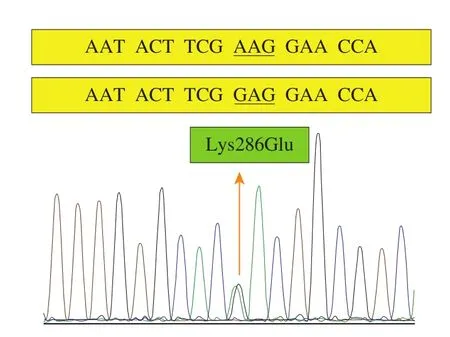
图5 菌株P2-81的Lys286定点突变测序对比Fig. 5 Sequence comparison of site-directed mutations at Lys286 of mutant P2-81
3 结 论
本研究采用易错PCR技术对海洋细菌Tamlana sp. S12中具有代表性的酶组分Alg-2进行体外定向进化。研究发现采用9 mmol/L Mg2+、0.1 mmol/L Mn2+的浓度组合可以使得易错PCR的突变率保持在合适范围。经过2 轮易错PCR及96 孔板发酵筛选,分别获得2 株正突变和2 株负突变菌株,其酶活力分别是亲本(2 675.2 U/g)的162%、241%、53%、11%。酶的动力学分析显示,正突变株P2-81的Km值比亲本降低50%,而负突变株N2-47的Km值比亲本提高106%,表明突变株对底物的亲和力有极大的上升和下降。基因测序及氨基酸序列分析结果表明,Glu、Thr、Ser、Asp和Tyr对褐藻胶裂解酶活力提高起到正面的积极作用,而保守区域Lys的突变起到负面的消极作用。后续人工合成的定点突变则进一步对Asp和Lys的正/负调节作用进行证实。本研究中获得的酶活力最高的突变株P2-81,其比活力为6 420.8 U/g,虽然和目前美国Sigma公司商业化的产品(比活力>10 000 U/g)相比还有不小的差距,但本研究室将继续采用更多轮次的易错PCR技术对褐藻胶裂解酶Alg-2进行定向进化,进一步提高其酶活力。本研究有助于深入理解褐藻胶裂解酶的催化机制,为后续利用理性设计手段改造褐藻胶裂解酶提供理论参考。
