免疫检查点PD-1/PD-L1小分子抑制剂的研究进展
2019-03-08田季平周金培张惠斌
田季平,张 剑,周金培,张惠斌
(中国药科大学新药研究中心,南京 210009)
近几年来肿瘤免疫疗法已成为肿瘤治疗领域的焦点。美国免疫学家詹姆斯·艾利森(James Allison)和日本生物学家本庶佑(Tasuku Honjo)凭借“发现负性免疫调节治疗肿瘤的疗法”获得了2018年诺贝尔生理学或医学奖。与直接针对肿瘤细胞的传统治疗手段不同,肿瘤免疫疗法是利用人体自身免疫系统对肿瘤细胞进行杀伤。肿瘤免疫疗法在临床上不断取得的成功,使得肿瘤免疫疗法成为目前炙手可热的肿瘤治疗手段[1]。免疫检查点(例如PD-1、CTLA-4、TIM-3等)通路的活化会抑制T细胞的激活,防止人体免疫系统的过度激活,维持正常机体的免疫耐受,避免自身免疫疾病的发生。肿瘤通过使自身和一些淋巴细胞过度激活免疫检查点通路导致肿瘤免疫逃逸的发生。在这些免疫检查点当中,PD-1/PD-L1的过度激活对于肿瘤的发展起着至关重要的作用。在肿瘤免疫疗法当中,针对免疫检查点PD-1/PD-L1通路的阻断剂无疑是最为闪耀的“明星”。
自2014年来,已有2种PD-1单抗和3种PD-L1单抗药物经FDA批准上市,并且这些单抗药物在多种肿瘤的临床治疗中取得了突破性进展。许多肿瘤患者的生存期显著延长,并且部分患者得到完全缓解。国内外有多款PD-1/PD-L1单抗处于临床研究阶段,基于抗体的免疫治疗已成为一个重要的研究领域。虽然单抗药物的临床效果显著,但仍存在一些无法回避的问题。由于单抗半衰期较长,并且与靶点结合时间过久,这些单抗类药物能够导致严重的免疫相关不良反应(immune-related adverse events,irAEs)[2-3]。单抗药物生产工艺复杂,价格昂贵且不便于储存运输,普通患者只能望“药”兴叹。与单克隆抗体相比,小分子药物具有价格低廉、可口服给药、便于运输和储存、良好的膜通透性和非免疫原性等显著优势。因此,寻找PD-1/PD-L1小分子抑制剂成为目前新药开发的热点。本文对近年来PD-1/PD-L1小分子抑制剂的研究进展进行总结。
1 PD-1/PD-L1的生物学机制
1.1 PD-1/PD-L1结构与功能
程序性死亡蛋白1(programmed cell death protein 1,PD-1也称作PDCD1和CD279),是由基因PDCD1编码,288个氨基酸残基组成的Ⅰ型跨膜蛋白,属于B7-CD28受体超家族成员[4-5]。它的结构包括4个部分:免疫球蛋白可变区(IgV)、跨膜区、免疫受体酪氨酸抑制基序(immunoreceptor tyrosine-based inhibitory motifs,ITIM)、免疫受体酪氨酸转换基序(immunoreceptor tyrosine-based switch motifs,ITSM)[6]。其在骨髓细胞、树突状细胞、自然杀伤细胞(NK)、单核细胞、CD4-CD8-胸腺细胞、调节性T细胞、B细胞以及抗原呈递细胞等多种免疫细胞表面均有表达[7]。PD-1有两个配体分别为PD-L1(CD274)和PD-L2(CD273),它们分别是由290与270个氨基酸残基组成的Ⅰ型跨膜蛋白,同属于B7家族,并且具有37%的同源序列。PD-L1由IgV和IgC样胞外区、跨膜区、短的胞质尾区3个部分组成。PD-L1表达在抗原呈递细胞、非淋巴器官和多种肿瘤细胞上[8-9]。虽然PD-L1与PD-L2都是PD-1的配体,但PD-L2表达范围较窄而且主要表达在树突细胞、单核细胞等免疫细胞上,研究发现PD-L1在肿瘤的免疫逃逸过程中发挥着主要作用。
1.2 PD-1/PD-L1信号通路
在正常的生理条件下T细胞并不会大量表达PD-1,当T细胞长期暴露在抗原刺激下,才会引起PD-1表达上调。同时活化的T细胞会通过释放γ干扰素(TNF-γ)、白细胞介素等细胞因子进一步诱导其他一些细胞过度表达PD-L1。PD-L1与PD-1结合后导致PD-1胞内域的免疫受体酪氨酸抑制基序(ITIM)和免疫受体酪氨酸转换基序(ITSM)发生磷酸化,进而招募酪氨酸磷酸酶SHP-1和SHP-2[10]。这些磷酸酶能够将T细胞抗原受体(TCR)信号通路上的多个关键蛋白去磷酸化,抑制TCR下游的信号通路,例如PI3K/AKT/mTOR、RAS/MEK/ERK、c-Myc等,进而抑制相关基因的转录,阻碍T细胞细胞周期进展,以及相关蛋白的表达。这些将抑制T细胞的增殖分化及细胞因子的产生[11-13](图1)。这种调节机制能够防止T细胞被过度激活,使人体免疫系统保持对自身抗原的免疫耐受,并减轻免疫反应对周围正常组织的损伤。

图1 PD-1/PD-L1信号通路图
肿瘤细胞通过过度表达PD-L1,持续激活PD-1/PD-L1信号通路造成多种免疫抑制。目前这些机制大概分为以下3类:①促进肿瘤特异性T细胞的凋亡[14];②能够使外周和淋巴组织的T细胞转化为丧失功能的调节性T细胞(Treg)和“衰竭”性T细胞(TEX)[15-16];③抑制效应T细胞和初始T细胞的激活,并且表达在免疫细胞表面的PD-L1也能影响抗肿瘤的CD8+T细胞的反应[17-18]。通过这样的免疫逃逸机制,肿瘤细胞能够轻松躲过免疫系统识别和打击。
因此,阻断PD-1与PD-L1的结合可以逆转上述免疫抑制机制,将有助于提高机体的免疫系统杀灭肿瘤的能力,这也为阻断PD-1/PD-L1介导的肿瘤免疫疗法提供了可靠的理论基础[19]。
2 PD-1/PD-L1信号通路小分子抑制剂
2.1 基于多肽的PD-1/PD-L1抑制剂
2014年报道的首个多肽类hPD-1抑制剂AUNP-12,由印度的Aurigene公司与PierreFabre实验室共同开发,由于AUNP-12比单抗代谢半衰期更短,故能够有效控制免疫治疗相关不良反应(irAEs)事件的发生。在表达hPDL2的HEK293细胞与hPD-1的结合试验中,AUNP-12能够抑制PD-1与PD-L2的结合,EC50达到了0.72 nmol/L;在挽救外周血单个核细胞增殖试验中,EC50为0.41 nmol/L[20]。动物试验表明AUNP-12具有良好的抗PD-L1活性,能够有效抑制肿瘤细胞的生长和转移,并且AUNP-12还具有良好的安全性,在所有给药剂量中均未表现出明显的不良反应。该化合物的具体结构尚未披露,目前根据该公司所公开的相关专利推测AUNP-12可能的结构为化合物1[21-23]。
2015年Aurigene公司又开发了一类环肽类化合物。在小鼠脾细胞荧光染料增殖试验中,化合物2能够诱导小鼠脾细胞的增殖。在小鼠的黑色素瘤高转移株B16F10皮下移植瘤模型中,该化合物可显著抑制肿瘤转移,使肿瘤转移发生率下降54%[24-25]。化合物3和4在小鼠脾细胞增殖挽救试验表现不俗,在小鼠重组PD-L1存在下,向抗CD3/CD28抗体(1 μg/mL)刺激的小鼠脾细胞中加入浓度为100 nmol/L的待测化合物来评估化合物挽救小鼠脾细胞能力,化合物3和4分别达到95%和94%的挽救率[26-27]。

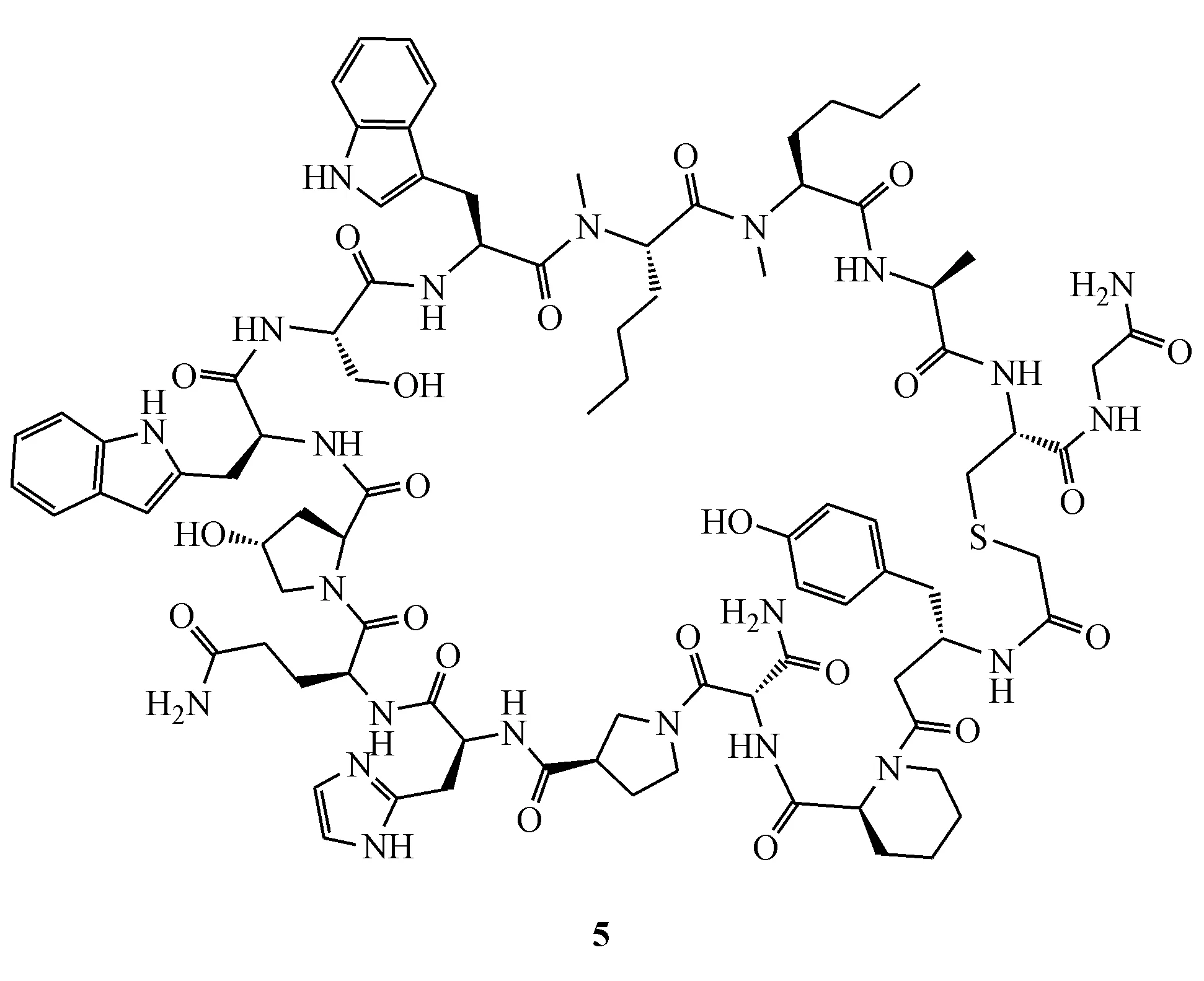
百时美施贵宝(BMS)公司开发了一类环肽PD-1/PD-L1抑制剂,这类抑制剂包含了13~15个氨基酸残基,其中化合物BMS-986189在2016年已进入Ⅰ期临床,适应证为脓毒症。化合物具体结构尚未披露,根据结构描述和相关专利推测BMS-986189可能为化合物5,在均向时间分辨荧光(homogeneous time-resolved fluorescence,HTRF)试验中,BMS-986189显示出对PD-L1有着极高的亲和性,IC50达到了1.03 nmol/L[28-32]。
清华大学刘磊课题组通过相位显示技术,利用噬菌体展示肽库筛选得到了由12个氨基酸组成的D型多肽DPPA-1,其氨基酸序列为:NYSKPTDRQYHF,它与PD-L1具有一定的亲和力,在与PD-L1的结合试验中,测得Kd为0.51 μmol/L,在CT26移植瘤小鼠模型中,该多肽能显著抑制肿瘤的生长[33]。2017年12月,中国科学院苏州纳米所朱毅敏课题组报道了一种可以靶向PD-L1的多肽TPP-1,TPP-1是由22个氨基酸残基组成的多肽,其氨基酸序列为SGQYASYHCWCWRDPGRSGGSK,与PD-L1结合的活性达到74 nmol/L。在大细胞肺癌H460细胞系的异种移植小鼠模型中,TPP-1能够显著增加IFN-γ的分泌与颗粒酶B的表达,并且使肿瘤体积明显减小[34]。
2.2 PD-1/PD-L1信号通路非肽类小分子抑制剂
2.2.1 磺酰胺类化合物 2011年,来自哈佛大学的Sharpe课题组筛选到一类磺胺间甲氧嘧啶和磺胺甲二唑衍生物,此类结构能够阻断mPD-1信号通路,其代表化合物为6、7和8。通过检测T细胞的激活状态及T细胞分泌IFN-γ的量来表征化合物对于mPD-1/mPD-L2的阻断效果,实验发现,这两类化合物在微摩尔浓度时就能够抑制mPD-1于mPD-L2的结合[35]。作为首次发现的PD-1小分子抑制剂,此类磺胺类衍生物有望作为先导化合物进行进一步改造和优化,设计具有成药性的化合物。

2.2.2 联苯类化合物 BMS公司在PD-1单抗与大分子生物药取得成功的同时,对PD-1/PD-L1信号通路小分子抑制剂领域进行了深入探索。2015年,该公司公开了第一篇关于联苯型免疫调节剂的专利。通过HTRF试验发现,此类化合物能够很好的阻断PD-1与PD-L1相互作用,部分化合物的蛋白活性达到纳摩尔级别,其中代表性化合物9和10的IC50分别为18和22 nmol/L[36]。同一年,在BMS公司公开的另外的一篇专利中,通过将化合物A部分的苯环用1,4-苯并二烷替换掉,并且在C部分的苯环上通过醚键引入间氰基苯,使化合物活性大幅提升,蛋白抑制活性普遍在0.6~10 nmol/L,代表性化合物11和12的IC50分别为2.25和1.4 nmol/L[37]。研究人员随后又对该类化合物进一步改造,在A部分疏水的联苯结构上通过一个碳链引入了一些亲水性的基团,化合物活性普遍得到提升,其代表性化合物为13和14,IC50分别为0.48和0.88 nmol/L[38]。在2018年,BMS公司披露了一类新的结构类型化合物,这类结构在原有基础上将左侧的结构替换成与右侧结构相同或相类似的结构,这样形成了成了一种呈“中心对称”的化合物,这类化合物的活性普遍小于1 nmol/L,其中代表性化合物15的IC50达到了0.04 nmol/L[39]。
波兰雅盖隆大学Holak课题组通过解析BMS专利化合物(化合物9)与PD-L1蛋白的晶体复合物发现,两个PD-L1蛋白形成了一个近似圆柱形的深疏水通道,而小分子如同“三明治夹心”一样位于该通道中间。如图2所示,化合物9通过诱导两个PD-L1二聚化,阻碍了PD-L1与T细胞表面PD-1的相互作用,由此起到阻断PD-1与PD-L1的相互作用[40-41]。除此之外,该课题组通过重组表达PD-1和TCR的Jurkat细胞充当T细胞,表达PD-L1和TCR配体的中国仓鼠卵巢细胞(CHO)充当刺激T细胞的抗原呈递细胞(APCs),当PD-1与PD-L1结合时重组Jurkat细胞不会产生免疫激活,所携带的荧光报告基因不能表达,当阻断PD-1/PD-L1信号通路时,Jurkat T细胞被激活,荧光报告基因表达。通过这种细胞试验可以测试BMS化合物直接阻断PD-1/PD-L1信号通路的能力,由此测得化合物11与化合物12的EC50分别为253和276 nmol/L,而已上市的单抗类药物例如pembrolizumab,nivolumab所测得的EC50分别为0.55和1.15 nmol/L[42]。


图2 化合物9诱导两个PD-L1蛋白二聚化[42]
研究证明,血清中游离的PD-L1蛋白(sPD-L1)浓度升高与肿瘤患者的不良预后有着密切的联系[43-44],并且血液中游离的PD-L1蛋白能够干扰血液中T细胞的激活[45-47]。其他研究也表明,sPD-L1能够与细胞膜上的PD-1结合[48]。Frigola等[49]发现sPD-L1能够诱导CD4+、CD8+T淋巴细胞细胞凋亡。有人推测sPD-L1能够与细胞膜上的PD-L1结合,能够阻止不同细胞间的PD-1/PD-L1结合,因此削弱了免疫系统的抗肿瘤作用。但是目前无法得知,sPD-L1是否是PD-1/PD-L1通路激活的主要因素。当前主流观点认为直接阻断膜结合PD-1/PD-L1的相互作用是抑制剂开发的主要策略,目前亟需验证此类小分子的成药性。
在国内,针对PD-1/ PD-L1通路的小分子抑制剂开发取得了显著进展。2017年中国医学科学院药物研究所冯志强课题组公开了一系列专利披露了一类溴代苄醚衍生物。此类化合物能显著阻断PD-1与PD-L1的结合,该类化合物与BMS所披露的化合物结构相似,结构上的区别为联苯结构中的甲基基团取代为溴。在HTRF试验中,多数化合物活性达到纳摩尔水平,其中部分化合物的IC50小于1×10-13mol/L,代表性化合物16和17的IC50分别为8.0×10-14和4.5×10-13mol/L。利用提取的人PBMC(人单核细胞),在anti-CD3/anti-CD28抗体激活T淋巴细胞的基础上,加入配体PD-L1抑制T淋巴细胞,考察待测化合物解除配体抑制作用的能力。实验结果显示,实施例化合物在10 nmol/L 时就能部分解除PD-L1对IFN-γ分泌的抑制作用。在小鼠的B16F10皮下移植瘤模型中,化合物18的钠盐在15 mg/kg剂量下,无论从肿瘤体积还是重量上,都可显著抑制皮下肿瘤的生长,其对肿瘤重量的抑制率达到了64.11%[50-52]。

2018年广州再极医药科技有限公司披露一类芳香乙炔或芳香乙烯类化合物。其代表性化合物19、20在HTRF试验中对PD-1/PD-L1相互作用有显著抑制作用,其IC50分别为18和48 nmol/L[53]。

2.2.3 其他杂环类化合物 Incyte公司通过将一些稠合杂环替代BMS原专利化合物的苯环,合成了许多新的小分子化合物,这些化合物可以阻断PD-1/PD-L1相互作用,代表性化合物为化合物21、22、23、24,在HTRF结合试验中发现许多专利化合物的IC50在10 nmol/L以下[54-55]。2018年10月Gilead公司所披露了一类阻断PD-1/PD-L1的小分子化合物,其中具有代表性的专利化合物25、26的IC50分别为0.068和0.051 nmol/L[56]。
临床前的体内数据表明CA-170能够显著解除PD-L1对T细胞的抑制,促进T细胞的增殖分化和IFN-γ的产生。在多种体内肿瘤模型中,CA-170显示出了与PD-1单抗类似的抗肿瘤活性。2016年Curis在美国开展了Ⅰ期临床试验,适应证为实体瘤和淋巴瘤,在Ⅰ期临床试验当中,CA-170显示出良好的药代动性质和安全性,并且显现出良好的抗肿瘤活性。
Curis公司于2017年12月在印度开展了Ⅱ期临床试验[Clinical Trials Registry of India (CTRI) registration no.-CTRI/2017/12/011026]。从2018年2月到2018年10月15日共有62名患者入组该临床试验,最后有37名患者参与评估,此次试验针对多种肿瘤(头颈鳞状细胞癌、非小细胞肺癌、高度微卫星不稳定性实体瘤和经典型霍奇金淋巴瘤)。从公布的临床数据来看,CA-170对于非小细胞肺癌和霍奇金淋巴瘤的效果最好,其整体临床获益率(Clinical Benefit Rate,CBR)分别为达到了70%与77.8%,具体数据如表1所示。其中一例霍奇金淋巴瘤受试者在400 mg/d的剂量下,连续60 d给药后肿瘤减小了57%,另外一例头颈癌受试者在400 mg/d给药剂量下,连续给药30 d后肿瘤减小了48%。比较之前PD-1单抗的临床数据,CA-170的400 mg剂量组的临床获益率与PD-1/PD-L1抗体相当[58]。
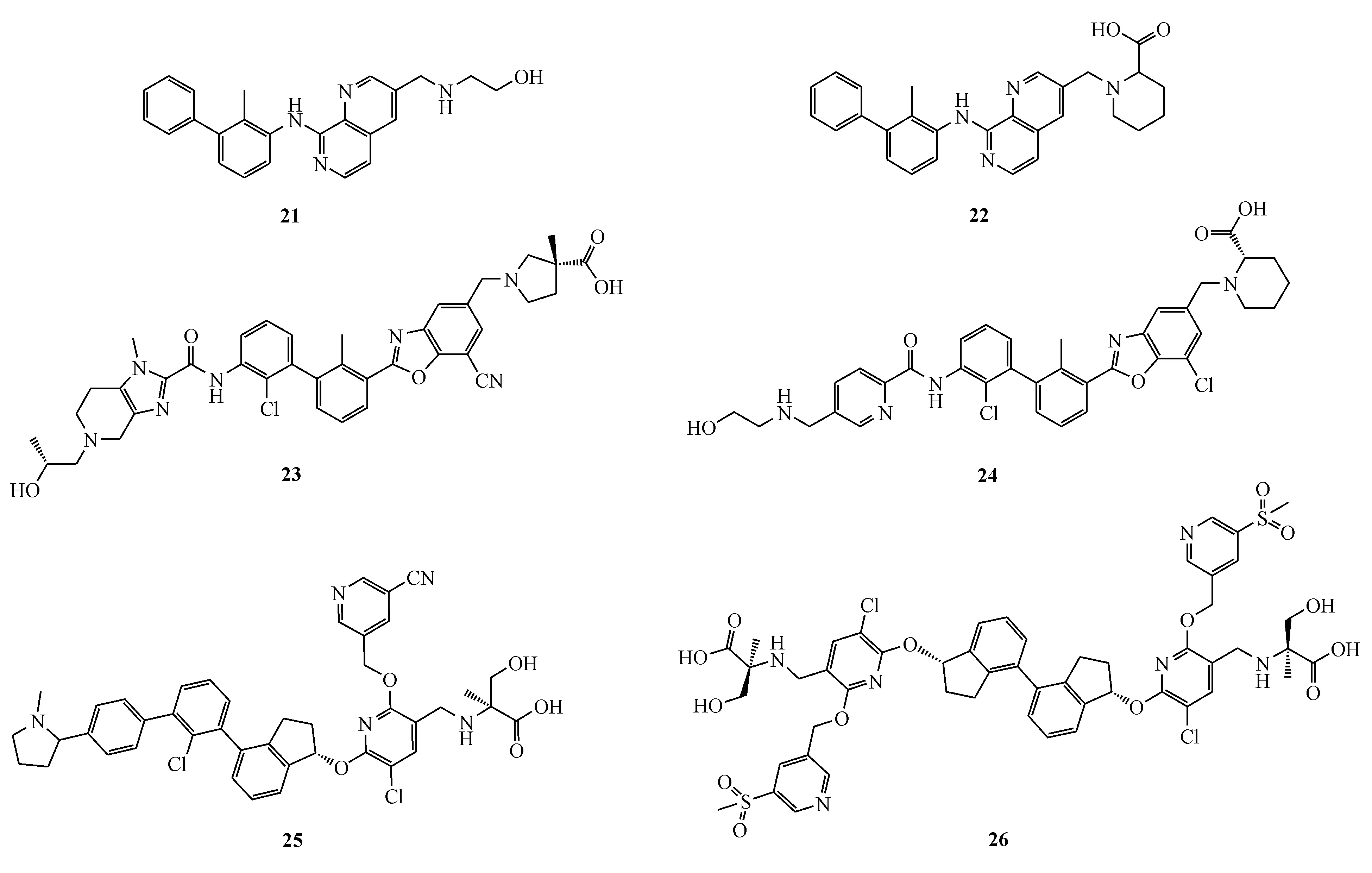

表1 CA-170Ⅱ期临床试验对部分肿瘤患者的临床获益率
值得注意的是,不论整体还是某一肿瘤类型,低剂量组(400 mg/d)的临床获益率要高于高剂量组(800 mg/d)的临床获益率。在体外的IFN-γ分泌实验和外周单核细胞挽救实验也发现了类似的现象,在低剂量时,解除免疫抑制效果随着药物浓度升高而升高,然后随着药物浓度增大,CA-170解除免疫抑制能力反而减弱。值得一提的是CA-170在安全性方面明显优于单抗,在高达1 200 mg/d的给药剂量下,也没有明显不良反应。虽然62例患者中出现了5例免疫相关不良事件(2例400 mg剂量和3例800 mg剂量),但在停药后各项指标恢复正常。而单抗药物在治疗中出现的免疫相关不良事件难以控制,即使停药后也不能逆转不良反应,并且需要使用大量的糖皮质激素和免疫抑制药物来治疗。

虽然已报道的PD-1/PD-L1小分子抑制剂的药性以及临床疗效还需进一步研究,但现阶段数据足以证明该类小分子化合物可阻断PD-1/PD-L1信号通路,所以开发PD-1/PD-L1小分子抑制剂可为肿瘤患者带来更多的福音。
4 结论与展望
自肿瘤免疫疗法诞生以来,肿瘤治疗领域有了许多突破性进展,特别是PD-1/PD-L1单抗药物的临床疗效显著。同时单抗类药物存在不可避免的缺陷,抗体类药物有较长的体内半衰期,加上本身具有免疫原性容易导致患者出现药源性免疫相关不良事件;另外大分子生物药由于通透性较差,体内分布不均,限制着生物药进入实体瘤的内部,导致疗效降低;生产难度大、治疗成本高昂、运输不便也是单抗类药物无法回避的缺点。虽然小分子抑制剂对靶标的结合活性低于抗体药物,但其可控制的药代动力学性质和成熟的研究体系,使其有可能克服抗体药物存在的问题。因此,设计合成阻断PD-1/PD-L1结合的小分子抑制剂具有十分重要的意义。
PD-1与PD-L1结合属于蛋白-蛋白相互作用,其接触界面具有高度平坦和疏水的特性,而且他们具有较大的相互作用表面和极少传统的小分子结合口袋,即使报道了相关晶体复合物,也很难找到适合小分子作用的位点,因此针对PD-1与PD-L1相互作用的小分子药物研发相对困难。虽然目前已有人源PD-L1与BMS公司小分子晶体复合物的报道,但该类小分子是通过诱导PD-L1二聚化间接阻断PD-1与PD-L1的结合,并不能直接阻断PD-1/PD-L1的结合。在体内是否能够实现细胞之间的PD-1/PD-L1的直接阻断作用需要进一步验证。当前小分子抑制剂研究还处于起步阶段,而且相关的活性评价体系和作用机制研究并不完善,解决这些问题是发展PD-1/PD-L1小分子抑制剂的当务之急。随着科学技术的发展,相信在不久的将来PD-1/PD-L1小分子抑制剂能够成功上市,造福广大的肿瘤患者。
