贵州武陵山地区19份柚资源的遗传多样性
2019-03-07王小柯郑乾明李文云柏自琴李金强
王小柯, 郑乾明, 罗 怿, 李文云, 柏自琴, 李金强
(贵州省农业科学院 果树科学研究所,贵州 贵阳 550006)
柚是一类重要的柑橘类果树,广泛分布于中国南方及东南亚地区。云贵地区作为内陆性系统柚的多样化中心之一,拥有极其丰富的地方柚类资源[1]。贵州省内柚资源主要分布于黔东北武陵山区,目前调查发现有酸柚、红肉柚、白柚等野生资源,经过长期的自然杂交和人工选育,这些野生资源形成了一系列具有优良性状的地方品种。当前对黔东北武陵山区柚资源尚无系统的研究报道,对其遗传多样性缺乏了解。
SNP位点是目前广泛应用的分子标记,具有分布广泛、分辨率高、共显性、易于高通量检测和分型等优点,可应用于遗传资源评价和遗传图谱构建等[2]。GBS技术是基于第2代测序技术大量快速挖掘SNP的简化基因组测序方法,主要利用酶切降低基因组复杂度,多样本高通量平行检测酶切位点附近的SNP,从而达到基因分型的目的。GBS分析具有同时检测多个样本,通量大、费用低的优点,广泛用于品种识别、遗传多样性评价和遗传图谱构建等[3-6]。笔者前期采集了贵州省武陵山地区6个区县共19份柚资源,分析其形态学特征发现,在叶形、花色、果肉色泽上均存在差异。因此结合GBS测序技术,从基因组水平上挖掘SNP位点,进行核苷酸差异分析及群体遗传多样性分析。拟通过简化基因组测序的方法,开发分子标记用于贵州地方柚品种及野生资源品种鉴定以及遗传多样性分析,以期为保护柚野生资源和地方品种资源、品种鉴定、挖掘优良性状奠定基础。
1 材料与方法
1.1 样品采集
通过实地考察和走访群众,在贵州省铜仁市6个区县共采集19份柚样品(表1)。采集时选择干净、无病虫害、幼嫩的春梢叶片。采集后用无菌水清洗2次并擦干,然后液氮速冻。
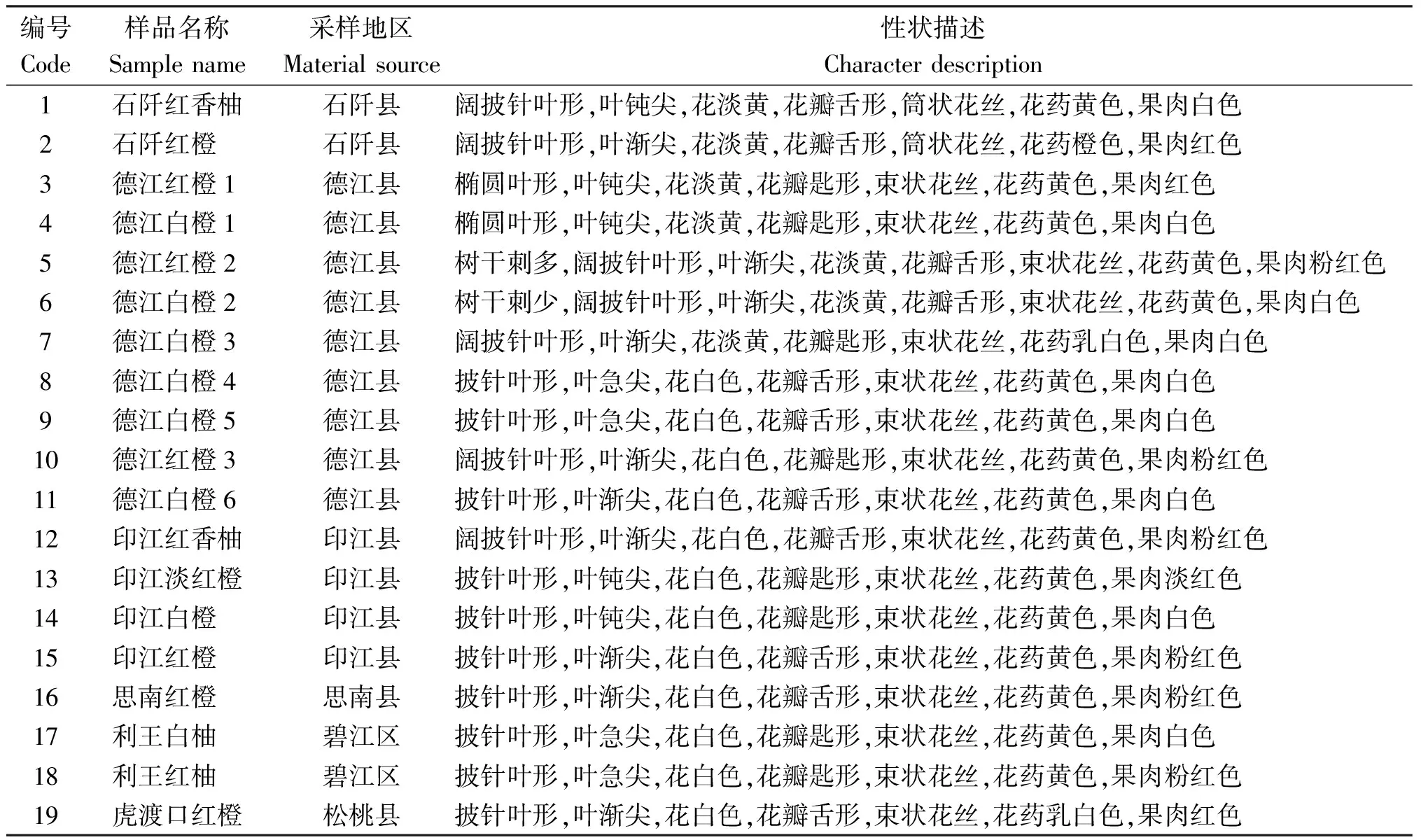
表1 武陵山地区不同区县19份柚样品的信息
1.2 基因组 DNA提取
19份柚样品叶片总DNA的提取使用植物基因组DNA提取试剂盒[天根生化科技(北京)有限公司,DP305],具体步骤参考试剂盒说明书。提取的DNA利用琼脂糖凝胶电泳检测其完整性,用Nanodrop 2000核酸测定仪检测浓度。检测合格的DNA样品送北京诺禾致源生物信息科技有限公司进行后续检测。
1.3 文库构建与测序
对柚参考基因组(http://citrus.hzau.edu.cn/orange/download/index.php)进行电子酶切预测,最终选择的限制性内切酶组合为MseⅠ+HaeⅢ+HaeⅡ。针对每个样品,首先用限制性内切酶组合对基因组进行酶切,酶切后加上带有条码的接头;接着对每个样品进行PCR扩增,将扩增后的所有样品混合;电泳后选择大小为240~290 bp的片段,利用 Illumina HiSeq 4000测序平台,进行双末端各150 bp测序,用于文库构建。
1.4 群体SNP检测
测序获得的原始数据,需要筛选以获得高质量序列,筛选条件:去除原始测序序列中的接头序列,去除单端测序中含N数量超过该序列长度10%的序列,去除单端测序中低质量(质量值Q≤5)碱基数超过该序列长度50%的序列。产生的高质量序列利用BWA(Burrows-Wheeler Aligner)程序与柚参考基因组比对(http://citrus.hzau.edu.cn/orange/download/index.php)。利用Samtools软件进行SNP检测,利用贝叶斯模型检测群体中的多态性位点。然后过滤掉测序错误率>1%的SNPs,过滤掉两者距离在5 bp范围内的SNPs,选择覆盖深度在平均深度的[1/3,5]倍的SNP,最终获得高质量的SNP。使用ANNOVAR软件对过滤后的SNP检测结果进行注释。
1.5 系统进化树构建及核苷酸多样性(π)分析
基于每个SNP位点在19份样品中的分型结果,采用邻接法(neighbor-joining methods)构建系统进化树。两两个体和之间的p-距离(Dij)通过如下公式计算:
式中,L为高质量SNPs区域长度,dij表示2个个体为不同基因型值的赋值表。
运用Treebest-1.9.2软件计算距离矩阵,以此为基础,通过邻接法(neighbor-joining method)构建系统进化树,使用FigTree软件生成系统进化树。引导值(bootstrap values)经过1 000次计算获得。
核苷酸多样性(π)是两两序列中具有不同核苷酸的比例。计算公式:
式中,pi是序列i的频率,其中pj是序列j的频率,πij是序列i与序列j的差异比例。使用软件ngs Tools软件(Fumagalli et al.,2014)进行计算。以200 kb长度为滑动窗口,100 kb为步长,进行核苷酸多样性(π)分析。
2 结果与分析
2.1 样本测序统计与质量评估
由表2看出,19个柚样本的原始测序总碱基为5.93 Gbp,去除低质量的序列后,产生高质量序列碱基为5.61 Gbp。19个样品测序碱基为255.11~388.38 Mbp,平均每个样本碱基数为311.76 Mbp。19个样品的Q20值为94.87%~96.28%,平均为95.76%;Q30值为87.44%~90.72%,平均为89.47%;GC含量为36.91%~37.62%,平均为37.3%,略高于柚参考基因组GC含量(34.99%)。
由表3可知,19个柚样品的比对匹配率为97.56%~98.78%,平均为98.47%。测序深度范围为9.39~12.54,平均为10.67。序列覆盖度范围为12.27%~13.84%,至少有1个碱基的平均覆盖度为13.03%,至少有4个碱基的平均覆盖度为7.24%。

表2 样本GBS测序的数据统计

表3 与参考基因组比对的匹配程度
2.2 群体SNP的筛选及分布
利用 Samtools软件检测后共获得608 466个SNP位点,过滤后共获得79 825个高质量的SNP位点。由图1可见,所有SNP位点在柚基因组9条染色体上基本均匀分布,分布密度为223.48~273.34个SNPs/Mbp。约5.52%(4 406个)的SNP位点未能有效定位到9条染色体上。对所有SNP位点在基因组上的位置统计表明(表4),52.19%(41 674个)位于基因间区域,14.19%位于基因的上下游,20.19%位于内含子区域,13.38%位于外显子区域,其中包括同义突变类型5.93%,非同义突变类型7.28%。
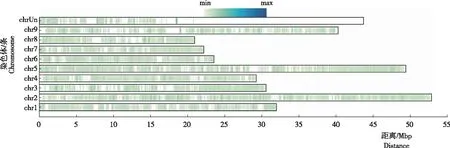
图1 SNP位点在柚基因组染色体上的分布
Fig.1 Distribution of SNP sites in chromosomes ofC.maximagenome
表4柚基因组的SNP检测及注释结果
Table 4 SNP detection and annotation result ofC.maximagenome

注(Note):外显子(Exonic):变异位于外显子区域;内含子(Intronic):变异位于内含子区域;剪接位点(Splicing):变异位于剪接位点(内含子中靠近外显子/内含子边界的2 bp);上游(Upstream):基因上游1 kbp区域;下游(Downstream):基因下游1 kbp区域;上游/下游(Upstream/Downstream):基因上游1 kbp区域,同时也在另一基因的下游1 kbp区域;基因间区(Intergenic):变异位于基因间区。
2.3 群体的系统进化树
由图2可见,19份样品被聚类为3个主要的类群。A类群中大部分是采集于德江县的柚子品种,另外还有少部分石阡县、思南县和印江县的品种。这一类群又可分为3个亚类群,A1类群包括样品1(石阡红香柚)、样品2(石阡红橙)、样品8(德江白橙4)、样品9(德江白橙5);A2类群包括样品5(德江红橙2)、样品6(德江白橙2)、样品7(德江白橙3)、样品11(德江白橙6)、样品16(思南红橙);A3类群包括样品10(德江红橙3)、样品14(印江白橙)、样品15(印江红橙)。B类群包括样品3(德江红橙1)、样品4(德江白橙1)、样品13(印江淡红橙)。C类群包括样品12(印江红香柚)、样品17(利王白柚)、样品18(利王红柚)、样品19(虎渡口红橙)。
从进化树可以看出,每个类群中的样品基本分布在同一地理区域,一些变异材料与其亲本也紧密聚类在一起,如样品5(德江红橙2)与6(德江白橙2)、3(德江红橙1)与4(德江白橙1)。样品6是在样品5上发现的果肉呈现粉红色的芽变枝条,样品3是在样品4上发现的果肉呈现粉红色的芽变枝条,进一步验证了结果的可靠性。
2.4 群体的核苷酸多样性
从图3可知,19份柚样品的群体核苷酸多样性值主要分布在4.84×10-6~4.0×10-4,最大值为1.5×10-3,平均值为1.09×10-4。

图2 19份柚资源的系统进化树
Fig.2 Phylogenetic tree of 19C.maximaresources
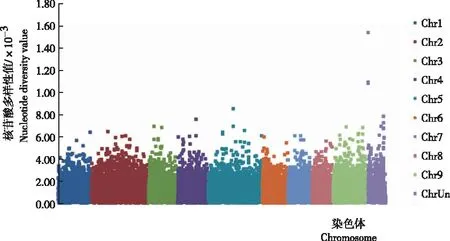
注:数值以200 kb为滑动窗口,100 kb为步长进行计算。
Note: These values were calculated in 200 kb sliding windows with 100 kb steps.
图319份柚资源基因组的核苷酸多样性
Fig.3 Nucleotide diversity of genome of 19C.maximaresources
3 结论与讨论
柚多为单胚,且长期以实生繁殖,因而极易产生实生变异和天然杂种,目前对柚类种质资源的演化关系及品种间系统关系尚缺乏了解。陈鹏等[7]采用花粉形态学分析方法研究湖南省地方柚类分类和遗传多样性;刘勇等[8]则是结合SSR 引物和AFLP分子标记对110份柚类基因型、12份野生近缘种进行遗传多样性研究;陈巍等[9]利用SRAP标记对13份浙南柚类地方资源和琯溪蜜柚及芽变材料进行遗传多样性分析和鉴定。近年来,随着高通量测序技术的发展,利用分子标记的方法对柚类进行遗传多样性研究变得越来越普遍,其中SSR和SNP 是广泛使用的分子标记。而SNP 标记将成为未来基因型鉴定的主要标记类型[10]。基于第二代测序技术的GBS技术因其成本低、高效且不受参考基因组限制等优点,逐渐成为一种快捷有效的SNP标记开发手段[11]。
与常规SSR标记比较,GBS分析在获得的标记数量上远远超过SSR标记[12-14]。并且,GBS分析获得的标记在染色体上均匀分布,具有更高的分析精密度,这是SSR标记不可比拟的。同时,这种优势也体现在试验所需周期和成本上。因此,结合高通量测序和SNP标记优点的GBS技术,在未来必将广泛用于资源多样性评价相关研究。笔者利用GBS测序分型获得了79 825个高质量SNP位点,这些位点广泛分布于基因间区、内含子和外显子中,在柚基因组9条染色体上均匀分布。群体核苷酸多样性值主要分布在4.84×10-6~4.0×10-4,平均值为1.09×10-4,遗传多样性较低,可能与群体的样本构成和选用的测序手段有关。利用这些SNP位点的分型结果构建系统进化树,19份柚样品被聚为3个主要类群,每个类群中的样品基本分布在同一地理区域,一些变异材料与其亲本也紧密聚类在一起,说明基于GBS测序分型的结果是准确可靠的。
研究中的柚群体核苷酸多样性值主要分布于4.84×10-6~4.0×10-4,远低于前人研究的柚群体19个样品的核苷酸多样性值(主要分布在1.0×10-3~2.0×10-3)[15],说明19份柚样品的遗传多样性较低。推测有以下2个方面的原因:首先是群体的样品构成,试验所用的19份样品采自贵州武陵山地区6个区县,尽管在植物学特征上存在差异,但样品的采集范围较窄;其次是检测的基因组范围不同,这与试验使用的技术策略和目的不一致有关。笔者采用的GBS基因分型技术,属于简化基因组测序,目的在于获得一些具有多态性的SNP位点,因此仅对酶切位点附近的SNP进行了检测,最终仅覆盖约13.03%的柚基因组,且试验成本较低;WANG等[15]利用了全基因组重测序技术,目的在于评价整个基因组的核苷酸多样性,其测序深度较高,基本覆盖了整个基因组,检测的位点更全面丰富,试验成本也相对较高。
德江红橙1(样品3)和德江红橙2(样品5)均是实地调查中发现的果肉呈现粉红或大红色的变异材料,分别采自其亲本德江白橙1(样品4)和德江白橙2(样品6)树上出现的枝条变异株。在系统进化树中,样品3与样品4、样品5与样品6分别聚在一起,其遗传差异极小,这也证实了2份变异材料确实分别来自其亲本上的枝条变异。这2对材料遗传背景极为相似,仅在果肉色泽上表现差异,是研究果肉色泽变异机理和色素代谢调控的良好材料。对这2对材料的差异SNP进行了统计和注释(结果未展示),仅约6.8%的差异SNP位点位于编码区。从注释结果中也未发现这些位于编码区的差异SNP与类胡萝卜素代谢途径基因相关,推测可能与采用的GBS技术所覆盖的基因组范围(仅覆盖13.03%的基因组)和遗传变异类型(仅检测SNP)有关。因此,下一步将利用这2对材料,从全基因组水平探讨色泽变异的遗传机理,为解析果肉色泽变异和色素代谢调控提供研究基础。
