一例Bjornstad综合征患儿BCS1L基因突变检测
2019-03-07郑璐瑶陈付英潘超兰程茹虹姚志荣
郑璐瑶 陈付英 李 越 潘超兰 程茹虹 姚志荣 李 明
Bjornstad综合征(Bjornstad syndrome)是一种罕见的先天性遗传性疾病,其遗传模式为常染色体隐性遗传,该病的致病基因是BCS1L基因。该病又称为扭曲发综合征,其典型的临床表现主要为先天性的头发扭曲以及感音神经性耳聋[1]。该研究中我们对一例Bjornstad综合征患儿及其父母进行了基因突变检测,患儿父母及双方家人表型均正常,现将结果报道如下。
1 对象与方法
1.1 对象 患者,女,6岁。患儿自出生即发现头发异常,头发扭曲、无光泽,易断裂,并出现脱发,随着年龄的增长,头发扭曲及头发脱失的表现逐渐加重(图1a~c),眉毛、睫毛亦受累,均稀疏(图1d)。显微镜下可见头发部分变扁平,有多处狭窄。患儿家长诉患儿左耳听力减退,右耳听力中度丧失。父母非近亲结婚,家族中无其他成员有类似疾病。体格检查:一般情况良好,身高、体重、智力发育正常。头颅无畸形,神经系统检查无异常发现。心肺肝脾未见异常,四肢肌张力正常。血、尿常规及血生化均正常。性激素、生长激素水平也基本正常。血清微量元素(铜锌铁钙镁铅)检测均在正常范围内。
耳鼻喉科检查:左耳轻度听力损失,右耳咽鼓管功能异常,高频听力下降。皮肤科检查:头发明显稀疏,呈扭曲状,干燥,可见断发、短发。眉毛、睫毛均稀疏。全身皮肤正常未见皮损。无牙齿、黏膜、指甲、汗腺的异常。
1.2 外周血基因组DNA抽取 在取得患儿家长的知情同意后,抽取患儿及其父母共3人的外周静脉血各2 mL,同时采集了100名正常对照的外周血,样本来自于上海交通大学医学院附属新华医院健康医生体检的剩余血样,该检测通过了新华医院医学伦理委员会的批准,并在充分告知后获得了知情同意。应用QIAampDNA Blood Minikit(德国QIAGEN公司)试剂盒提取基因组DNA。
1.3 二代靶向测序和Sanger测序 采用二代靶向测序包检测患儿的突变基因,对于二代靶向测序发现的突变位点,应用Sanger测序对患儿及其父母进行验证。
2 结果
测序结果显示,患儿在BCS1L基因发现两处复合杂合突变,在第7个外显子上发现了新的c.818delC缺失突变,导致p.A273fs,即在273位氨基酸处丙氨酸变成终止氨基酸,产生了一个截短蛋白,该突变遗传自患儿的父亲;在第8个外显子上发现c.917G>A错义突变,导致p.R306H(精氨酸突变为组氨酸),该突变遗传自患儿的母亲。以上2个BCS1L基因突变在100名正常对照个体中未检测到,提示它们为该病的致病变异,而非单核苷酸多态。

图1 a~c:头发扭曲及头发稀疏;d:眉毛、睫毛稀疏,缺如
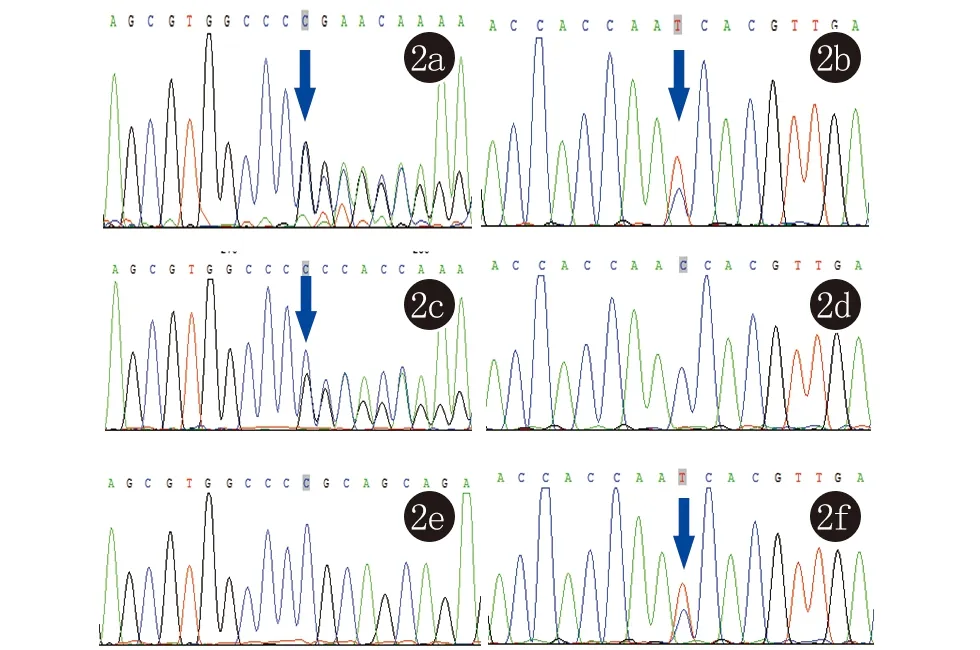
a:患儿c.818delC缺失突变;b:患儿c.917G>A错义突变; c、d:患儿父亲测序图;e、f:患儿母亲测序图
图2 BCS1L基因测序图
3 讨论
Bjornstad综合征是一种极其罕见的先天性遗传性疾病,又称扭曲发综合征。1965年,Bjornstad首次报道该病[1],临床上主要表现为先天性头发扭曲和双侧感音神经性耳聋[2],患者通常在出生后不久即出现头发扭曲并随着年龄增长而加重,头发干燥,无光泽且易于折断,毛干部分多处扁平,头发易在扭转处形成结,常在结处断裂,并形成局部或弥漫性脱发,睫毛、眉毛亦可受累。患儿还可出现严重程度不同的听力损失。曾经报道过该病出现的一些伴随症状[3],如智力发育迟缓等,但是发病率是未知的。
该病临床表现为扭曲发的可鉴别疾病有Menkes病[4],Crandall综合征[5],外胚层发育不良等。其中Menkes病是X连锁隐性疾病,铜代谢障碍;Crandall综合征只在男性患者中报道过,其临床表现不仅包括扭转发和神经性耳聋,还可出现性腺功能减退;而外胚层发育不良,则可出现头发、牙齿、指甲、汗腺的异常。本研究中患儿血清铜、性激素、生长激素均在正常范围内,且未发现其他异常,所以可以排除这些疾病。
1998年,Neto等[6]将Bjornstad综合征的致病基因定位于2q34-36。2007年,Hinson[7]将致病基因定位于D2S2210和D2S224之间物理距离为2Mb的区域,并对该区域的44个基因进行了DNA测序,发现Bjornstad综合征是由BCS1L基因突变所致。本研究通过二代靶向测序包,我们发现该患儿的突变基因是BCS1L基因。BCS1L基因编码线粒体内膜的一种蛋白质,包括419个氨基酸。Bjornstad综合征的突变位点位于BCS1L的AAA(与ATP酶相关的各种细胞活动的缩略词)区域的外表面[8],BCS1L基因突变导致线粒体功能异常[11],通过破坏线粒体呼吸小体(人类线粒体呼吸的基本单位)的组装,从而导致毛发和听力病变。以往报道,BCS1L基因突变还可以导致2种致命的综合征,一种为线粒体复合物III缺乏症[9],表现为新生儿肾小管病变和脑病与肝衰竭。另一种为CRACILE综合征,表现为宫内发育迟缓,氨基酸尿,胆汁淤积,铁超载,乳酸中毒和早期死亡[10]。
本文报道的患者父母否认近亲结婚史,患儿携带复合杂合突变,分别为第7个外显子的缺失突变c.818delC和第8个外显子的错义突变c.917G>A,其中前者突变来自于父亲,突变导致基因编码框移突变,即p.A273fs*27,会导致移码和蛋白水平的提前终止密码子,推测可能导致该基因所编码的蛋白功能缺失,从而导致疾病的发生。该突变在目前的文献以及数据库中未被报道。后者突变来自于母亲,即p.R306H,精氨酸突变为组氨酸,该突变最初是在挪威家系报道过的[7],功能研究已经表明p.R306H的错义突变可干扰线粒体呼吸小体的组装,导致病变的发生。
迄今为止,已经报道了10余例Bjornstad综合征的BCS1L突变位点。我们的研究进一步扩展了Bjornstad综合征的致病基因BCS1L突变谱,对于进一步了解BCS1L基因在线粒体的致病作用提供依据。通过基因检测明确患儿的基因诊断,能够提高皮肤科医生的临床诊断水平,有助于患儿疾病预后的评估,同时指导患儿的父母再生育时要行植入前产前基因诊断或产前基因诊断。
