IgG4相关性肾病的病理改变及特征
2019-03-06综述审校
王 荣 综述 唐 政 审校
IgG4相关性肾病(IgG4-related kidney disease,IgG4-RKD)是IgG4相关性疾病(IgG4-related disease,IgG4-RD)所有肾脏累及的统称,是一种免疫介导的炎性纤维化疾病,其病理特征为受累组织内大量IgG4+浆细胞浸润、席纹状纤维化,糖皮质激素治疗多有效[1]。IgG4-RKD可表现为肾小管间质炎症(IgG4-related tubulointerstitial nephritis,IgG4-TIN)、膜性肾病(IgG4-related membranous nephropathy,IgG4-MN)及其他肾小球性病变、腹膜后纤维化、肾脏炎性假瘤、慢性硬化性肾盂炎、肾动脉炎,其中以IgG4-TIN最为常见,其次为IgG4-MN[2]。本文重点就IgG4-TIN及IgG4-MN的病理特征作一综述。
流行病学
IgG4-RKD好发于中老年男性,平均年龄65岁[2]。目前尚无大样本的临床数据资料提供回顾性研究,故人群的确切发病率和患病率尚不明确。有代表性的主要是日本对23例IgG4-RKD患者和梅奥诊所对35例IgG4-RKD患者的研究,在Saeki等[3]研究的23例患者中男性20例(87%),年龄40~83岁,平均年龄65.2岁。在Raissian等[4]研究的35例患者中男性30例(86%),年龄20~81岁,平均年龄65岁。近期日本肾脏病学会(Japanese Society of Nephrology,JSN)公布了一项2012~2013年47家医疗机构的 6 978 例肾活检样本数据,发现共有47例IgG4-RKD患者,近15年来年龄>40岁的日本人群中IgG4-RKD发病率是0.9~3.1/100万[5]。
临床表现及病理特征
大体标本特点IgG4-RKD病变不仅仅局限于肾皮质,也常累及肾髓质、肾包膜及腹膜等。大体标本可见双肾实质肿胀,占位样改变,肾积水(多由输尿管受累、腹膜后纤维化或包块压迫引起),受累部位肉眼呈白色,质硬[6]。病变部位与非病变部位间界限清楚,此为IgG4-RKD特征之一;淋巴浆细胞可浸润至肾包膜外形成突出于肾脏表面的单发或多发病灶,是该病的另一特征[7]。
IgG4-TIN IgG4-TIN可有少量至中等量蛋白尿,个别患者偶有镜下血尿,肾小管损伤标志物(如尿N-乙酰-β-D-葡萄糖苷酶、尿视黄醇结合蛋白、尿白介素18等)常升高,肾功能损害较常见[8]。
IgG4-TIN病理表现可呈局灶或弥漫性,有时会形成瘤样肿块[9]。其典型的病理特征为肾间质大量浆细胞及淋巴细胞浸润(图1A)、席纹状或鸟眼样纤维化。炎症细胞浸润及纤维化主要位于肾皮质及血管周,肾髓质相对少见[6]。炎症细胞浸润范围内可见肾小管萎缩,肾小管狭窄及小管囊样扩张[10],部分肾小管损毁,甚至仅残留基膜结构,残存肾小管间距增宽,部分肾小管基膜(TBM)增厚,肾小管区域可有轻度灶性单核细胞性小管炎、浆细胞性小管炎及嗜酸性粒细胞性肾小管炎[9]。受累间质偶见Mott细胞,即细胞质中含有球形免疫球蛋白包涵体的浆细胞[11]。有时可见较多嗜酸性粒细胞浸润,此时需与药物相关性急性间质性肾炎相鉴别,后者多有明确的用药史,常见白细胞尿、白细胞管型及严重的小管炎。Masson三色染色可见TBM有免疫复合物沉积[11]。肾小球一般正常,少见闭塞性脉管炎,可能与细针穿刺肾活检取材较小、有一定局限性相关。Hara等[6]在5例尸检病例的肾组织中均发现了闭塞性静脉炎,佐证肾活检取材的局限性。Raissian等[4]将IgG4-TIN间质纤维化分为三种类型:(A)急性间质性肾炎,伴少量纤维化;(B)广泛间质纤维化,伴炎症细胞浸润;(C)少细胞性纤维化,并建议由纤维化类型初步推测实际患病病程,即A型病程较短,而C型病程较长。Yoshita等[12]分析了IgG4-TIN和非IgG4-TIN的光镜下特点,建议尚未常规开展IgG4免疫病理染色的单位可以将下述特点作为两者鉴别的依据:IgG4-TIN病灶多区域性分布、边界清楚,间质纤维化呈鸟眼样或席纹状,纤维化严重程度常与患者肾功能不相符,炎性细胞浸润可突破肾包膜,常见TBM免疫复合物沉积;而中性粒细胞浸润、重度小管炎、肉芽肿性损害、坏死性脉管炎多支持非IgG4-TIN的诊断。
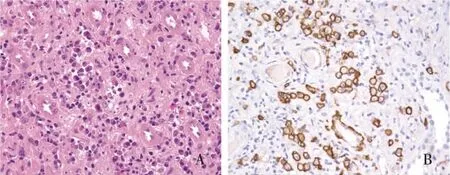
图1 A:肾小管间质大量淋巴浆细胞浸润(HE,×400);B:肾小管间质大量IgG4+浆细胞浸润(IH,×400)
约85%的IgG4-TIN患者免疫荧光下可见弥漫性或局灶性TBM免疫复合物颗粒状沉积,以IgG4为主,部分患者伴有κ链、λ链及C3的沉积,但IgM和C1q不多见。免疫复合物沉积的区域与TIN所累及的区域大致相对应,且与IgG4-TIN间质纤维化类型有关:A型无TBM免疫复合物沉积,B型中95%有TBM免疫复合物沉积,C型均有TBM沉积[4]。如若免疫荧光发现IgG4阳性浆细胞浸润,则高度提示IgG4-TIN可能,在与其他原因导致的肾小管间质性肾炎相鉴别时具有重要意义。
电镜下可见无定形电子致密物沉积于TBM,位置与免疫荧光下TBM沉积物部位一致,主要分布于间质炎性细胞浸润部位[4]。IgG4-TIN肾小球内通常无电子致密物沉积。
IgG4-MN 当IgG4-RKD累及肾小球时可表现为大量蛋白尿或肾病综合征,以膜性肾病最多见。Alexander等[13]报道了9例IgG4-MN的患者,发现所有受试者均呈现肾病范围的蛋白尿,其中5例伴有TIN,8例行肾活检的患者肾组织抗磷脂酶A2受体抗体染色均阴性,提示该8例患者膜性肾病为IgG4-RD继发而非原发。IgG4-MN光镜下肾小球大致正常或毛细血管袢增厚,上皮下、内皮下及系膜区可见免疫复合物沉积,肾小球基膜钉突形成,肾衰竭者可见球性硬化[2]。Saeki等[7]报道7%~10%的IgG4-TIN合并有IgG4-MN,但两者之间的相互联系有待进一步研究。
IgG4-MN免疫荧光可见肾小球基膜上IgG4、C3、κ链及λ链的颗粒样沉积,仅少量IgGl和IgG3沉积,抗磷脂酶A2受体抗体染色阴性[14]。与IgG4-TIN相比,IgG4-MN中TBM免疫复合物沉积少见,仅占33%[15]。临床上需注意与抗磷脂酶A2受体抗体相关膜性肾病相鉴别,后者的抗原主要是内源性的足细胞抗原,免疫复合物沉积部位主要在上皮下,肾组织抗磷脂酶A2受体抗体染色阳性;而约33%的IgG4-MN患者肾小球内皮下及系膜区亦可见免疫复合物沉积[13],肾组织抗磷脂酶A2受体抗体染色阴性[16],且多有IgG4-RD其他器官累及的表现。
电镜下IgG4-MN可见许多电子致密物在上皮下沉积。
IgG4相关其他肾小球病变IgG4相关肾小球病变还可表现为IgA肾病、系膜增生性病变、膜增生样病变、毛细血管内增生性病变等其他病理类型[17],伴或不伴IgG4-TIN。
IgG4相关血管病变血管病变在IgG4-RKD中较少见。Sharma等[18]报道了一例浆细胞性肾动脉炎的IgG4-TIN患者,病变主要累及中小动脉,可见内膜、中膜和外膜有大量淋巴浆细胞浸润,动脉壁中大量IgG4+浆细胞浸润,未见动脉纤维素样坏死及弹性纤维断裂,病变血管中无嗜中性粒细胞浸润。Hara等[6]观察到5例IgG4-TIN尸解病例肾皮质可见中小血管周围炎症或纤维化。有时可以在IgG4-TIN患者中看到闭塞性静脉炎[15]。
免疫表型特征
肾组织大量IgG4+浆细胞浸润是IgG4-RKD的特征性表现(图1B)。JSN于2011年提出IgG4-RKD诊断标准(表1),其中将肾组织大量IgG4+浆细胞浸润的界值设定为炎症细胞密集区IgG4+细胞计数>10个/高倍视野,或IgG4+/IgG+浆细胞比例>40%,该界值的敏感性为100%,特异性为91%[19]。但有学者提出部分IgG4-TIN患者IgG4+/IgG+浆细胞比例<40%[11],甚至有学者报道了两例有浆细胞浸润但无IgG4+浆细胞的IgG4-TIN患者[20-21]。Mizushima等[22]提出IgG4-RD受累的组织不同(如肾、下颌下腺、皮肤等),IgG4+细胞数目及IgG4+/IgG+浆细胞比例的界值亦不同。此外,大量IgG4+细胞浸润并非IgG4-RKD所特有,也可见于狼疮性肾炎、ANCA相关性血管炎、新月体性肾炎、特发性间质性肾炎、肉芽肿性血管炎、糖尿病肾病等[23],故浆细胞浸润是诊断IgG4-RKD的必要条件,但缺乏特异性,尚需结合患者的临床表现、影像学等才能做出最后诊断[24]。

表1 日本肾脏病学会提出的IgG4-RKD诊断标准[19]
a:临床病理学上要排除以下疾病,如肉芽肿性血管炎、Churg-Strauss综合征、髓外浆细胞瘤等;b:影像学上要排除以下疾病:恶性淋巴瘤、肾癌、肾梗塞、肾盂肾炎、肉芽肿性血管炎、结节病、肾转移癌等
IgG4-RKD不同部位的纤维化成分不同,皮质区主要成分为fibronectin、胶原Ⅳ和串珠样分布的胶原Ⅵ,胶原Ⅰ及胶原Ⅲ染色阴性;而血管旁则主要为胶原Ⅰ及胶原Ⅲ。IgG-RKD早期即有α-SMA阳性的肌成纤维细胞浸润,并随疾病进展逐渐减少,提示产胶原Ⅰ、Ⅲ、Ⅳ的肌成纤维细胞在IgG4-RKD早期纤维化过程中起重要作用[6]。
除此之外,有研究表明IgG4-RD患者T辅助细胞2(Th2)及调节性T细胞(Treg)显著增多,提示该两种细胞在IgG4-RD发病中起重要作用[25-26]。Th2诱导嗜酸性粒细胞、肥大细胞增多及血清IgE增加,促进生发中心形成,刺激IgG4的产生和IgG4阳性浆细胞的浸润[27]。Treg分泌的白细胞介素10(IL-10)促进B细胞分化为浆细胞并产生IgG4,同时,Treg分泌的转化生长因子β1(TGF-β1)激活成纤维细胞,诱导内皮细胞及上皮细胞分化为肌成纤维细胞,促进组织纤维化[28-29]。故可考虑免疫染色Th2、Treg来源的细胞因子或趋化因子(如IL-10、TGF-β1等),进一步探讨IgG4-RKD的免疫特性。Kawamura等[30]对16例IgG4-RKD、16例干燥综合征(Sjögren syndrome,SS)、17例原发性间质性肾炎(ITIN)进行IgG、IgG1、IgG4、CD38、CD3、CXCR3、CCR4、Foxp3、胶原Ⅰ、Ⅲ、Ⅳ、Ⅵ、TGF-β1免疫组化分析,发现IgG4-RKD肾间质浆细胞、嗜酸性粒细胞浸润及纤维化程度明显重于SS及ITIN,IgG4+浆细胞/IgG+浆细胞、Foxp3+细胞/CD3+细胞、TGF-β1+细胞/浸润细胞数均高于SS及ITIN,且IgG4-RKD中Foxp3+细胞/CD3+细胞与IgG4+浆细胞/IgG+浆细胞、TGF-β1+细胞/浸润细胞数呈正相关,Foxp3+细胞和TGF-β1+细胞可共定位,TGF-β1+细胞/浸润细胞数与纤维化程度正相关,提示Treg促进IgG4产生,Treg分泌的TGF-β1在IgG4-RKD间质纤维化中起重要作用。
诊 断
目前国际上尚无统一的IgG4-RKD诊断标准,但达成共识的是,组织学对于诊断IgG4-RKD至关重要。对于IgG4-TIN,具有代表性的诊断标准主要有JSN[19]及梅奥诊所[4](表2)提出的两个诊断标准,这两个诊断标准均要求先排除新月体性肾炎、ANCA相关性血管炎、系统性红斑狼疮、冷球蛋白血症、肉芽肿性血管炎、感染、肿瘤等疾病[9]。前者设立的诊断标准严格,特异性高,但易漏诊;后者相对宽泛,在诊断非典型及早期IgG4-TIN时敏感性更高,但特异性低,误诊率增加[11]。值得注意的是,JSN提出的诊断标准中血清IgG4升高为确诊所必需,而Kamisawa等[31]提出血清IgG4的测定可作为筛选项目,但不能作为诊断标准之一,因为约20%的IgG4相关自身免疫性胰腺炎患者血清IgG4正常,而约4-10%的健康人会出现血清IgG4升高,故血清IgG4敏感性及特异性尚需进一步探讨。两个诊断标准制定依据的病例数均较少,需要临床进一步验证。国内学者Tang 等[14]对480例间质性肾炎患者进行回顾性筛查,纳入疑似IgG4-TIN病例12例,通过对其血清学、影像学、组织学及其他脏器的损害等情况进行综合评估,确定该12例患者中有4例为新月体肾炎,4例为IgA肾病,3例为间质性肾炎,1例为狼疮性肾炎,再根据IgG4阳性浆细胞的分布规律以及临床特点,结合梅奥诊所及JSN所建议的诊断标准,最终确定只有2例为IgG4-TIN。可见现行诊断标准有待进一步完善。
治疗及预后
IgG4-RKD是IgG4-RD的肾脏累及,其治疗原则与IgG4-RD的治疗原则基本一致。2015年IgG4-RD国际诊疗指南共识指出:糖皮质激素是IgG4-RD诱导缓解的一线药物(94%同意);因部分患者单用激素不能有效控制疾病,故该类患者在起始治疗时可予糖皮质激素联合免疫抑制剂治疗(46%同意);复发患者可再使用糖皮质激素治疗,维持缓解期应使用免疫抑制剂(81%同意)[32]。IgG4-RKD大多数情况下对激素有效,但并非所有的IgG4-RKD患者经激素治疗后肾功能均能好转,少数表现为进行性肾功能衰竭的患者快速进入终末期,从而需长期肾脏替代治疗。吗替麦考酚酯、硫唑嘌呤、甲氨蝶呤、他克莫司及环磷酰胺等免疫抑制剂也用于该病的治疗,但目前多为个案报道,尚需临床试验进一步明确其疗效[33]。利妥昔单抗清除B细胞,有学者提出可用于IgG4-RKD难治性病例,以达到替代/减少激素剂量的目的[34-35]。

表2 梅奥诊所提出的IgG4-TIN的推荐诊断标准[4]
a:必备条件,伴有其他影像学、血清学检查或其他脏器受累中至少1条即可诊断;TIN:肾小管间质炎;TBM:肾小管基膜
小结:IgG4-RKD好发于中老年男性,临床表现及病理类型多样,以IgG4-TIN最为常见,其次为IgG4-MN。特征性病理表现为大量IgG4+浆细胞浸润、席纹状纤维化。肾脏病理对于IgG4-RKD的诊断尤为重要,免疫病理在探讨其发病机制中起重要作用。大部分IgG4-RKD病例对糖皮质激素治疗敏感,延误诊疗将增大肾衰竭的风险。
