血红蛋白H检测在α-珠蛋白生成障碍性贫血和MDS诊断中的意义
2019-03-05薄丽津赵玉平
孙 雪,薄丽津,徐 艳,赵玉平
(中国医学科学院北京协和医学院血液病医院/血液学研究所检测中心,天津 300020)
血红蛋白H(HbH)为一种异常血红蛋白,患者父母一方a珠蛋白基因型为(α-/αα),另一方a珠蛋白基因型为(--/αα),患者自父母双方继承了异常a珠蛋白基因,其基因型为(α-/--),由于α链不足,无α链配对的β链自行聚合成β四聚体即HbH。通常情况下,HbH是诊断α-珠蛋白生成障碍性贫血又称α-地中海贫血(简称α-地贫)的金标准。工作中,在其他非α-地贫患者的标本中也可检测到HbH。本文总结本院 2007年1月至2017年10月19 699例HbH检测结果,对其阳性结果结合病例资料及相关文献报道做一回顾性分析。
1 资料与方法
1.1一般资料 所有标本均来自临床上怀疑有溶血发生的患者。2007年1-10月本院检测中心溶血室检测HbH 19 699例,阳性结果79例,阳性率0.4%。同一期间临床上有明确诊断病例39例。其中α-地贫组34例,男性11例,女性23例,男女比例为1∶2.09,年龄3~60岁,中位年龄21.5岁;骨髓增生异常综合征(MDS)伴获得性HbH病组5例,男性4例,女性1例。
1.2仪器与试剂 分别采用Sysmex XE-5100型血细胞分析仪及配套血常规及网织红细胞检测试剂;Beckman Coulter AU2700型全自动生化分析仪及配套血清胆红素检测试剂;D-10血红蛋白分析仪(美国Bio-Rad公司)及配套血红蛋白组分分析试剂盒;北京瑞尔达生物科技有限公司提供的游离血红蛋白测定试剂盒。
1.3方法
1.3.1血常规及网织红细胞检测 静脉血以乙二胺四乙酸二钾(EDTA-K2)抗凝后,使用Sysmex XE-5100型血细胞分析仪检测红细胞计数(RBC)、血红蛋白(Hb)、网织红细胞百分数(Ret)、红细胞平均体积(MCV)等指标。
1.3.2血清胆红素检测 空腹静脉血经促凝、离心后,取上层血清,使用Beckman Coulter AU2700型全自动生化分析仪检测总胆红素(TBIL)、间接胆红素(IBIL)等指标。
1.3.3HbH、血浆游离血红蛋白、血浆结合珠蛋白检测 静脉血以EDTA-K2抗凝后,使用D-10血红蛋白分析仪进行Hb组分分析,对怀疑有HbH的标本再用酸性血红蛋白醋酸纤维膜电泳确认,具体方法参见文献[1]。空腹静脉血经肝素钠抗凝、离心后,取上层血浆,采用过氧化物酶法测定游离血红蛋白(F-Hb);用电泳法检测血浆结合珠蛋白(Hp),具体方法参见文献[1]。
1.4统计学处理 采用SPSS17.0统计软件进行数据处理,对α-地贫组各标本的HbH含量及相对应的血常规、网织红细胞、胆红素、F-Hb进行Pearson相关性分析,P<0.05为差异有统计学意义。
2 结 果
2.1α-地贫组 实验室检查结果结果见表1。本研究对各标本的HbH含量及相对应的血常规、网织红细胞、胆红素、F-Hb进行两组数据间的Pearson相关性分析,结果见表2。
2.2MDS伴获得性HbH病组 共5例,男性4例,女性1例。5例患者的一般资料及实验室相关检查结果见表3、4。

表1 34例α-地贫患者实验室检查结果
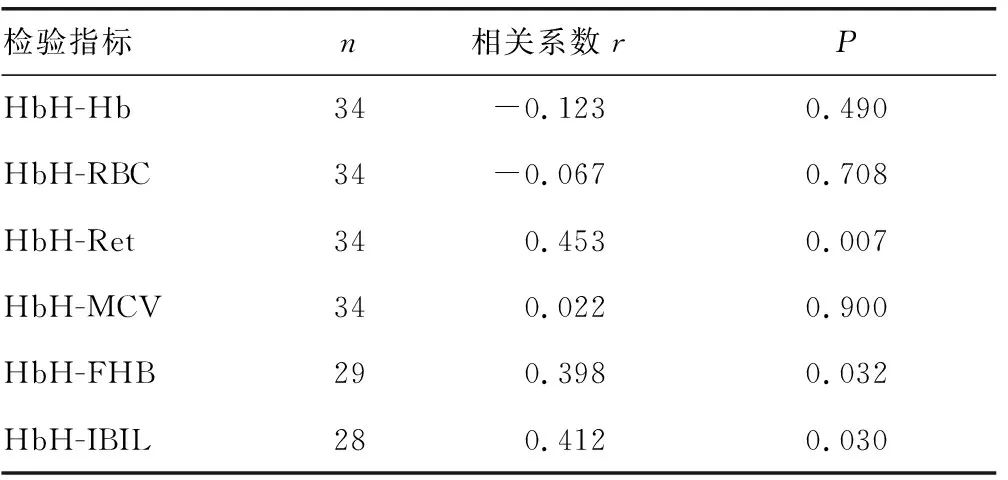
表2 实验室检查的相关性分析
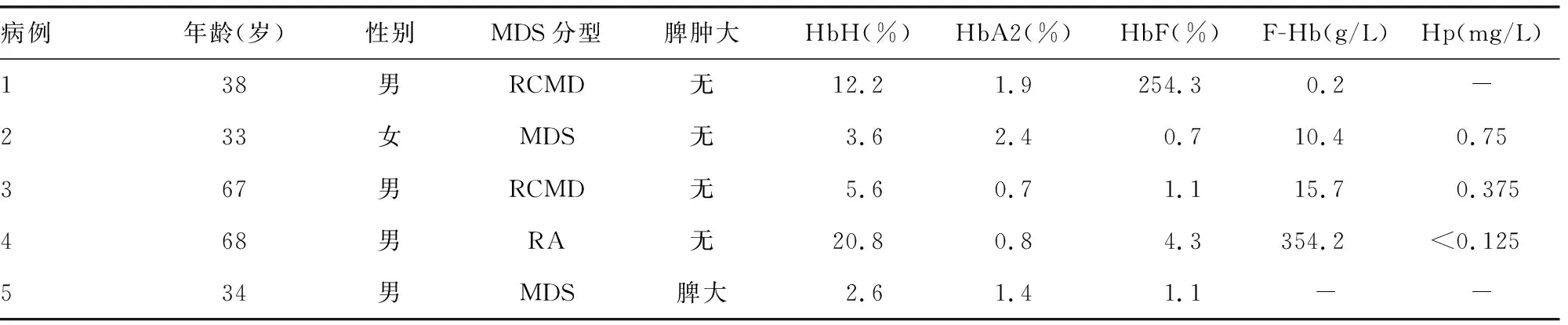
表3 5例MDS伴获得性HbH病患者一般资料及相关溶血检查结果
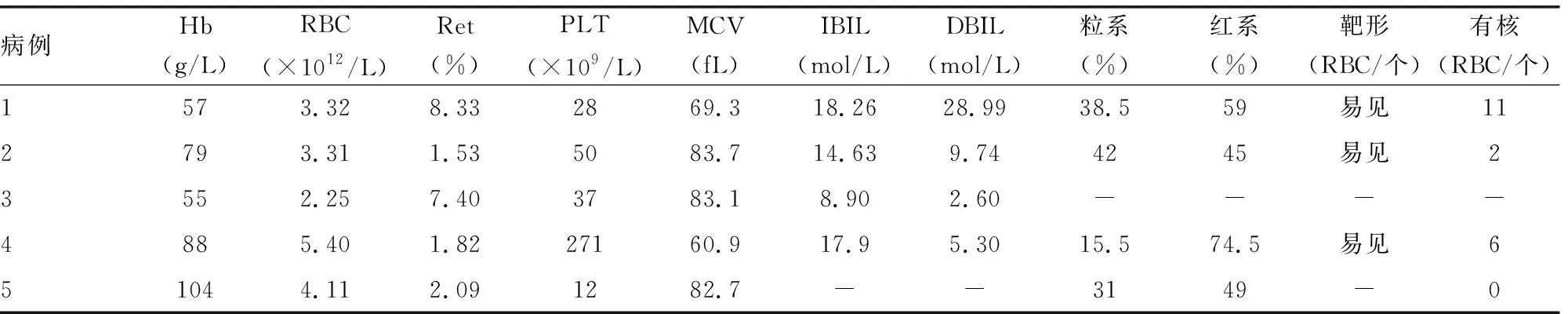
表4 5例MDS伴获得性HbH病患者血常规、生化及形态学检查结果
3 讨 论
3.1HbH病 HbH病是一种由于血红蛋白α基因缺失导致血红蛋白合成异常的α-地贫。α-地贫是一组由于α珠蛋白链的合成受到部分或完全抑制所引起的遗传性溶血性贫血。α珠蛋白基因定位于第16号染色体短臂,健康人自父母双方各继承两个α珠蛋白基因(αα/αα),合成足够的α珠蛋白链。若继承了父母有缺陷的α珠蛋白基因,则使α珠蛋白生成障碍而发生α-地贫。
不同类型的α-地贫临床表现与α基因受累的数目和程度有关。HbH病为缺乏3个α基因所致。在HbH病患者体内,由于α链合成的不足,患者在胎儿期或成人后出现过剩的γ珠蛋白链或β珠蛋白链,形成γ四聚体即HbBart′s或β四聚体即HbH。表现在Hb电泳中出现HbH,其含量约为5%~40%。本研究中27例患者的HbH含量在此范围中,与文献报道相符[2],7例不在此范围中。
HbH氧亲和力高、不稳定、易在红细胞内沉淀形成HbH包涵体,导致红细胞变形性降低,易在体内破坏,发生溶血性贫血。 所以这类患者一般情况下游离血红蛋白升高,结合珠蛋白降低,间接胆红素升高。本研究对各标本HbH含量与对应的RBC、Hb、Ret、MCV、FHB及IBIL的检测结果进行了相关性分析,结果表明HbH含量与Ret,FHB及IBIL的检测结果呈正相关。本文中34例患者,29例(85.2%)患者Ret随HbH含量增加而升高;27/29例(93.1%)患者FHB随HbH含量增加而升高;23/28例(82.1%)患者IBIL随HbH含量增加而升高。与文献报道相符[2]。
3.2MDS伴获得性HbH病 个别MDS和急性髓系白血病(AML)等恶性血液疾病患者也可出现HbH,因主要见于MDS,并且由于其发病机制的独特性,STEENSMA等[3]提出“MDS相关α地中海贫血(ATMDS)”。ATMDS诊断标准:(1)存在HbH;(2)患有其他恶性血液病(主要是MDS);(3)排除先天性HbH病。
本研究的5例MDS伴获得性HbH病患者,结合临床表现、血常规及骨髓象改变,确诊为MDS。同时,5例患者HbH检测阳性,但依据患者均系祖籍北方及病史和家族史,可以排除遗传性HbH病。最后诊断为“MDS伴获得性HbH病”。
现已证明MDS出现HbH有两种机制:一种为MDS异常克隆的红系祖细胞4个α珠蛋白基因中有3个缺失,即--/α-,α珠蛋白链合成减少;另一种是由于ATRX基因突变所致,该突变使所有4个α珠蛋白基因表达明显降低[4-5]。研究结果显示大部分ATMDS患者存在体细胞X-linked地贫伴智力障碍(ATRX)基因点突变[6-7],仅有少数研究报道ATMDS患者存在第16号染色体a珠蛋白基因区域异常。ATRX基因异常通过多种途径影响a珠蛋白的表达,这与ATRX蛋白功能密切相关。作为染色质重塑因子[8-10],ATRX蛋白异常将影响组蛋白H3.3嵌入、基因稳定性及修复等过程[11],引起ATMDS患者α珠蛋白基因异常及DNA复制缺陷;RATNAKUMAR等[12]研究结果显示,抑制性组蛋白变体mH2A作为ATRX蛋白新伴侣,亦参与调节α-地贫的表现型,ATRX蛋白可抑制mH2A在染色质的沉积;ATRX蛋白异常时,mH2A在α珠蛋白基因位点聚集程度增加,从而抑制α珠蛋白表达。不论何种原因所致α珠蛋白表达受阻,均会在血红蛋白电泳中出现HbH,造成部分MDS患者HbH检测阳性。
4 结 论
虽然HbH是诊断α-地贫的金标准,但HbH不仅仅存在于α-地贫中,在MDS也可检测到。给临床诊断提供了依据。同时本文探讨了HbH病患者的HbH含量与Ret,F-Hb及IBIL的检测结果呈正相关,而MDS伴获得性HbH病的病例较少无法进行统计学分析,有待于样本量扩大开展进一步研究。
