2018年FDA批准的新药评述(Ⅰ)
2019-03-01魏利军
魏利军
(舒泰神生物制药股份有限公司,北京 100176)
美国FDA是全世界经验最成熟、最权威、效率最高的药品审评机构;与此同时,美国也是全球第一大药品市场,因此FDA常被视作新药从实验室走向市场的第一道大门。 2018年,FDA批准了59个新分子实体,其中14个被授予突破性疗法,43个获优先审评通过。这些药物中,约有10个产品有望成为重磅炸弹药物(即上市后第5年的销售额不低于10亿美元的产品),其中最被看好的产品分别为bictegravir/恩曲他滨/丙酚替诺福韦(Biktarvy)、tezacaftor/ivacaftor (Symdeko)、apalutamide(Erleada)、erenumab(Aimovig)、cannabidiol(Epidiolex)、elagolix sodium(Orilissa)、lanadelumab(Takhzyro)和 cemiplimab(Libtayo)。为了更好地解释获批新药对未来市场的影响力,本文使用了科睿唯安预测数据进行阐释,感谢科睿唯安的数据支持。
1 177Lu-Dotatate
2018年1月26日,FDA批准了Advanced Accelerator Applications公 司 的177Lu-dotatate( 商 品 名:Lutathera,1),用于生长抑素受体阳性的胃肠胰神经内分泌瘤(GEP-NET)治疗。Lutathera是一种放射性镥(177Lu)标记的生长抑素类似物,可与生长抑素受体结合,尤其是对亚型2受体具有很高的亲和力。药物与该靶点阳性的肿瘤细胞结合后,镥释放出β射线,进而破坏肿瘤细胞。一项有229例晚期生长抑素受体阳性GEP-NET患者参与的临床试验(NETTER-1)数据显示:与长效奥曲肽单独治疗组相比,本品联合长效奥曲肽治疗组患者中位总生存期(OS)显著延长,分 别 为 NR(31.0,NE)vs27.4(22.2,NE)(NR即not reached,未达到;NE即not evaluate,未评估),风险比为0.52[1]。神经内分泌肿瘤是一类起源于胚胎的神经内分泌细胞、具有神经内分泌标记物和可以产生多肽激素的肿瘤[2],每年发病率为1/27 000[3]。除了神经内分泌瘤,本品还在开发嗜铬细胞瘤(Ⅱ期临床)、小细胞肺癌(Ⅰ/Ⅱ期临床)等适应证,部分分析师认为本品有销售额峰值超过10亿美元的潜力。

2 Bictegravir/恩曲他滨/丙酚替诺福韦
2018年2月7日,FDA批准了吉利德的Biktarvy用于1型人类免疫缺陷病毒(HIV)感染治疗。Biktarvy是一种三联疗法,含新一代整合酶抑制剂bictegravir(BIC,2)、核苷类逆转录酶抑制剂恩曲他滨(FTC)和丙酚替诺福韦(TAF)[4]。相比吉利德前一代抗HIV药物(“鸡尾酒”疗法),新型整合酶抑制剂bictegravir无需再使用肝药酶抑制剂cobicistat来延长作用时间,并以丙酚替诺福韦替代替诺福韦二吡呋酯,从而大幅降低了肾毒性和骨骼毒性。一项临床试 验 将 Biktarvy(BIC/FTC/TAF) 与 dolutegravir+恩 曲他滨+丙酚替诺福韦(DTG/FTC/TAF)组合疗法进行了抗病毒疗效对比,经过48周治疗,BIC/FTC/TAF治疗组病毒载量低于每毫升50拷贝的患者比例为92.4%(290/314),DTG/FTC/TAF组 为 93.0%(293/315),达到统计学意义上的非劣效性。BIC/FTC/TAF治疗组整体不良反应发生率明显低于DTG/FTC/TAF治疗组(26%vs40%),其中呕吐发生率分别为5%和17%[5]。由于“鸡尾酒”疗法需要长期用药,除了疗效和耐药性外,不良反应和便捷性也是影响产品成败的关键因素,基于这两方面的优势,本品有望成为艾滋病治疗市场的领头羊,科睿唯安预测本品在2024年的销售额将达到66.7亿美元,是2018年最值得关注的药物之一。
3 Tezacaftor/Ivacaftor
2018年2月12日,FDA批准了Vertex公司的组合疗法tezacaftor/ivacaftor(3)(商品名:Symdeko),用于F508del基因突变的纯合子,或囊性纤维化跨膜传导调节因子(CFTR)基因突变且有体外/临床证据显示对本品敏感的囊性纤维化(CF)患者的治疗。一项Ⅲ期临床试验数据显示:504名F508del基因突变的纯合子患者接受本品或安慰剂治疗24周,治疗组患者第1秒用力呼气容积占预计值百分比(pp FEV1)相比基线上升4%,安慰剂组则下降1%,达到主要治疗终点。此外,另有临床试验证明Symdeko相比tezacaftor单独治疗更有效[6]。CF是CFTR基因突变所引起的疾病,临床表现为慢性阻塞性肺病、胰腺功能不全及汗腺受累所致的汗液钠、氯异常升高。CF是一种在白种人中常见的致命遗传疾病,发病率达1/2 000,亚洲人和非洲黑人中较为少见[7]。CF治疗方案较为缺乏,本品相较lumacaftor/ivacaftor组合疗法(Orkambi),效果有所提升(pp FEV1:4%vs2.4%)[8],获得了FDA突破性疗法、孤儿药和优先审评等3项资格认定,并有望成为重磅炸弹药物,科睿唯安预测本品在2024年的销售额将达到19.90亿美元。
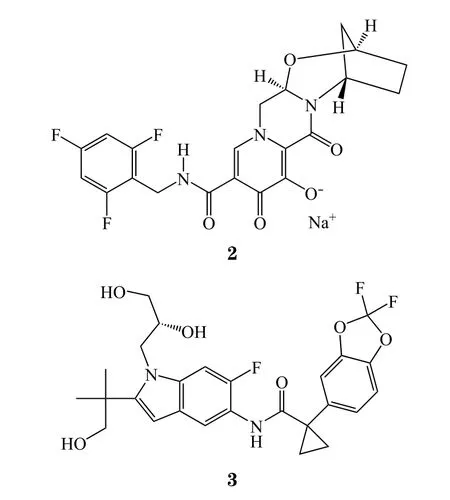
4 Apalutamide
2018年2月14日,FDA批 准 了 强 生 的apalutamide(商品名:Erleada,4),用于非转移性去势抵抗的前列腺癌(NM-CRPC)治疗。Apalutamide是一种雄性激素受体抑制剂,SPARTAN临床试验中,1 207名NM-CRPC患者在睾丸切除或促性腺激素释放激素(GnRH)类似物治疗的基础上,接受本品或安慰剂治疗,结果显示:本品治疗组患者无转移生存期中位值达40.5个月,而安慰剂组仅为16.2个月。前列腺癌是一种高发癌症,2017年约有16.12万美国人被诊断为前列腺癌,致死病例高达2.67万[9]。本品疗效出色,用药周期较长,而且有望获批用于转移性去势抵抗的前列腺癌,市场前景非常光明,是2018年最值得关注的新药之一,科睿唯安预测本品在2022年的销售额为15.70亿美元。
5 Ibalizumab
2018年3月6日,FDA批准了Taimed公司的ibalizumab(商品名:Trogarzo)与其他抗逆转录药合用,治疗高度耐药的HIV-1感染。Ibalizumab是一种人源化的IgG4单抗,可非竞争性结合CD4细胞,以阻碍HIV-1进入细胞内。在一项单组设计、开放标签的Ⅲ期临床试验中,40名多种抗逆转录药物治疗失败的严重耐药HIV-1感染者,在优化抗病毒治疗方案的背景治疗下添加本品治疗,结果显示:33名患者(83%)病毒载量相比基线(每毫升4.5log10拷贝)至少减少每毫升0.5 log10拷贝,平均病毒载量减少为每毫升1.1 log10拷贝。在第25周时,43%的患者的病毒载量小于每毫升50拷贝,50%的患者病毒载量小于每毫升200拷贝[10]。虽然大部分HIV-1感染者在多种抗逆转录药物的治疗下,病毒载量得到很好的控制,但部分患者在经过多种抗逆转录病毒治疗后出现高度耐药,医生束手无策,死亡风险较高;本品虽然潜在用药人群不大,但为医生提供了一种新的治疗选择[11]。市场方面,本品可能要走高价策略,但成为重磅炸弹药物的可能性很小,科睿唯安预测本品在2024年的销售额为2.93亿美元。
6 Tildrakizumab
2018年3月20日,FDA批准了太阳制药的tildrakizumab(商品名:Ilumya),用于适合接受光照或激光治疗的中重度斑块状银屑病患者的治疗。Tildrakizumab是一种人源化的IgG1单抗,靶点为IL-23p19,FDA批准tildrakizumab上市,是基于2项多中心临床试验,926名患者分别接受100 mg本品或安慰剂治疗,12周后,tildrakizumab治疗组银屑病皮肤面积与严重性指数(PASI)至少下降75%(PASI 75)的患者比例分别为64%和61%,安慰剂组仅分别为6%和6%;治疗组整体评价法(PGA)评分为0分或1分和至少提高2分的患者比例分别为58%和55%,而安慰剂组仅分别为7%和4%[12]。虽然银屑病是一个非常庞大的治疗市场,但竞争已经非常激烈,在强生的Tremfya(IL-23抗体)、礼来的Taltz(IL-17A抗体)和诺华Cosentyx(IL-17A抗体)等竞争对手面前,本品在疗效上处于劣势,甚至与阿达木单抗和阿普斯特相比,太阳制药都没有足够的把握,因此本品的销售额在5年内突破10亿美元的可能性不大,科睿唯安预测本品在2022年的销售额为1.2亿美元。
7 Burosumab
2018年4月17日,FDA批准了Ultragenyx公司的burosumab(商品名:Crysvita)上市,用于1岁及以上儿童和成人X连锁低磷血症(XLH)治疗。Burosumab是一种成纤维细胞生长因子(FGF)23抑制剂,本品的有效性已在4项临床试验中得到了确认。在这些试验中,94%的成人患者每月1次接受本品治疗后血磷值达到正常水平,而接受安慰剂治疗的患者只有8%达正常水平。在儿童患者中,高达94% ~ 100%的患者经过本品每2周1次的治疗后,血磷值达到正常水平。XLH是一种严重的罕见疾病,在美国约有3 000名儿童和1.2万成人罹患此病。XLH不同于其他形式的佝偻病,维生素D治疗无效,burosumab是该治疗领域的首个获批的药物,尽管成为重磅炸弹药物的潜力不大,但其上市具有突破性意义[13]。
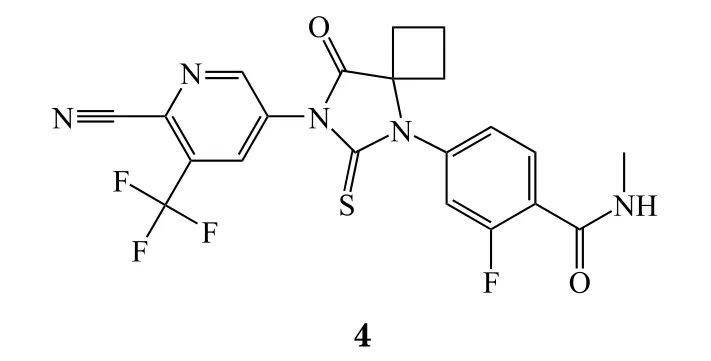
8 Fostamatinib disodium
同在2018年4月17日,FDA还批准了Rigel公司的脾酪氨酸激酶(Syk)抑制剂fostamatinib disodium(商品名:Tavalisse,5),用于慢性免疫性血小板减少症辅助治疗。FDA批准本品是基于2项安慰剂对照的临床试验(FIT-1和FIT-2)和一项开放标签的临床试验(FIT-3),在FIT1和FIT2试验中,47名先前接受过促血小板生成素受体激动剂(TPO-RA)治疗的患者,有8名(17%)对本品有稳定的应答[14]。因为fostamatinib潜在治疗对象不多,而且只有17%的人有稳定的应答,因此本品成为重磅炸弹药物的潜力不大,科睿唯安预测本品在2024年的销售额为4.23亿美元。阿片类药物危机愈演愈烈,阿片类药物戒断治疗已渐渐产生一个巨大的市场,US Worldmeds公司经过2项临床试验证明了本品的有效性,为美国阿片类药物戒断治疗带来全新的治疗选择,但成为重磅炸弹药物的可能性不大。
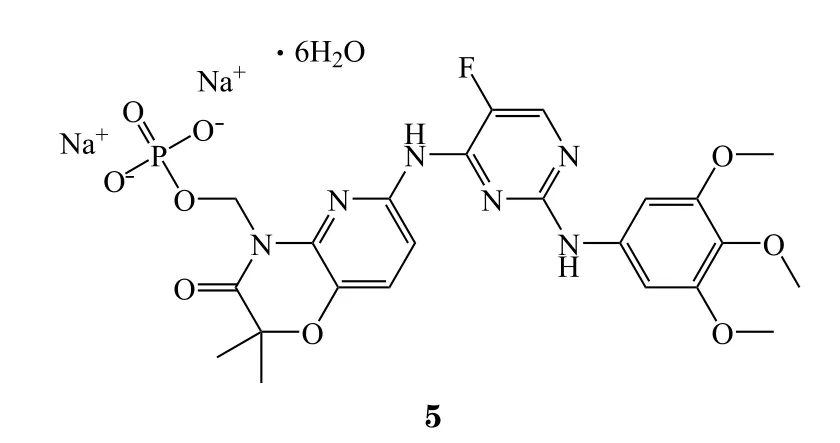
9 Fosnetupitant/帕洛诺司琼
2018年4月19日,FDA批准了Helsinn公司的复方静脉注射剂fosnetupitant(6)/帕洛诺司琼(palonosetron)(商品名:Akynzeo),联合地塞米松用于预防高致吐性化疗药引起的急性和迟发性恶心呕吐。本品由五羟色胺-3(5-HT3)受体拮抗剂帕洛诺司琼和神经激肽1(NK1)受体抑制剂fosnetupitant组成。Fosnetupitant可被视为奈妥匹坦的前药,通过体内水解后会转化为奈妥匹坦,随着磷酸基团的引入,水溶性大幅增强,为制备注射剂提供了必要的条件。由于奈妥匹坦和帕洛诺司琼组合疗法早在2014年就获得FDA批准,多项临床试验也已证明了奈妥匹坦和帕洛诺司琼组合疗法的有效性,故FDA并未要求Helsinn公司开展更多的临床试验来证明fosnetupitant的有效性[15]。对于化疗引起的剧烈恶心呕吐,注射或许是比口服更好的选择,虽然FDA此前已经批准了Heron公司的阿瑞匹坦静脉注射乳剂,但其配方中含有大量脂质,受众面将远不及Akynzeo注射液。
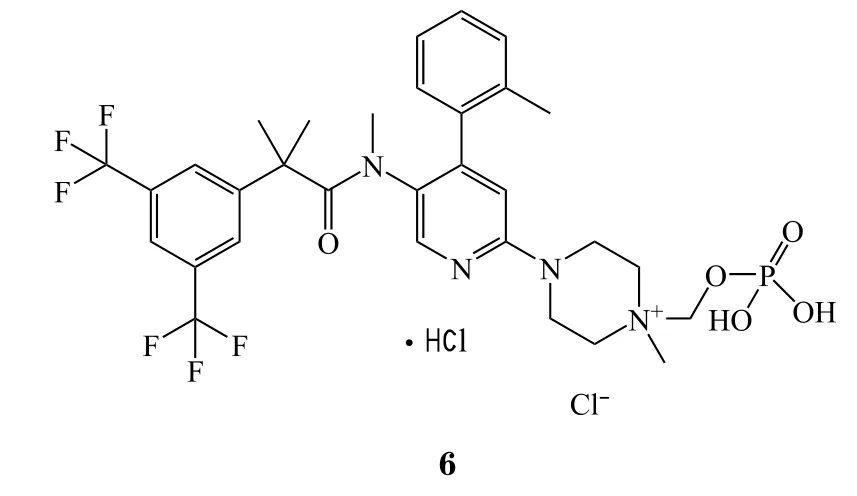
10 洛非西定
2018年5月16日,FDA批准了US Worldmeds公司的洛非西定(lofexidine,商品名:Lucemyra,7),用于减轻或解除阿片类药物的戒断综合征。洛非西定是一种肾上腺素α2受体激动剂,是首个被FDA批准用于阿片戒断综合征治疗的非阿片类药物[16]。洛非西定是近年来老药新批的典型案例,其于1990年已在英国上市[17],但美国一直没有批准该药。近年来,美国的

11 Erenumab
2018年5月17日,FDA批准了安进和诺华联合开发的erenumab(商品名:Aimovig),用于偏头痛治疗。Erenumab是首个获批上市的降钙素基因相关肽(CGRP)受体抑制剂,FDA批准该产品上市是基于3项临床试验的结果。在第1项试验(NCT 02456740)中,955名阵发性偏头痛患者分别接受本品70 mg每月1次、140 mg每月1次或安慰剂治疗,6个月后,70 mg每月1次治疗组患者每月偏头痛平均发作天数相比基线下降3.2 d,140 mg每月1次治疗组下降3.7 d,而安慰剂组仅下降1.8 d[18]。第2项试验(NCT 02483585)是一项有577名阵发性偏头痛患者参与的双臂临床试验,经过3个月的治疗,70 mg每月1次治疗组患者每月偏头痛发作天数相比基线下降2.9 d,安慰剂组下降1.8 d。第3项试验(NCT 02066415)是一项共有667名慢性偏头痛患者参与的临床试验,经过3个月治疗,70 mg每月1次治疗组患者每月偏头痛发作天数相比基线下降6.6 d,140 mg每月1次治疗组下降6.6 d,而安慰剂组仅下降4.2 d[19]。偏头痛是一种常见的慢性头痛,全球约有15%的人受这种疾病折磨,未满足的临床需求巨大[20]。虽然处在临床末期的CGRP受体抑制剂较多,但与本品相比,临床疗效并没有质的提升,相反,Aimovig因首发的优势而有望达到重磅炸弹药物级别,科睿唯安对本品在2024年的销售额预测值为12.45亿美元。
12 环硅酸锆钠
2018年5月18日,FDA批准了阿斯利康的环硅酸锆钠(sodium zirconium cyclosilicate,商品名:Lokelma),用于高钾血症治疗。本品是一种不吸收的硅酸锆,对钾离子有非常高的亲和力,可优先捕获钾以交换钠或氢。本品通过增加钾的粪便排出,以起到降低血钾的作用[21]。本品的有效性在3项临床试验中得到了确认,除了高钾血症,本品开发的适应证还有高氨血症,目前已经处在注册阶段。在众多高钾血症用药中,本品的疗效较为出色,是销售额峰值有望达到10亿美元的重量级产品之一。
13 Avatrombopag
2018年5月21日,FDA批准了Akarx公司的促血小板生成素受体激动剂avatrombopag(商品名:Doptelet,8),用于治疗慢性肝病导致的血小板减少症。ADAPT-1和ADAPT-2试验共纳入435名慢性肝病和严重血小板减少症的患者,考察了本品的安全性和有效性。试验中,受试者分别接受40 mg和60 mg的avatrombopag或安慰剂治疗,5 d后,2个剂量组患者的血小板计数相比基线均显著增加,而且与安慰剂存在显著性差异[22]。尽管本品获得了FDA孤儿药和优先审评2项特殊通道的资格,但潜在受众并不大,而且同靶点的药物还有诺华的eltrombopag和盐野义的lusutrombopag,因此成为重磅炸弹药物的潜力不大,科睿唯安预测本品在2024年的销售额为3.88亿美元。

14 Pegvaliase
2018年5月24日,FDA批准了Biomarin公司的pegvaliase(商品名:Palynziq),用于血液中苯丙氨酸浓度大于 600 μmol · L-1的苯丙酮尿症治疗。Pegvaliase是一种重组苯丙氨酸氨裂解酶的聚乙二醇修饰物,可将苯丙氨酸转化为氨和反式肉桂酸,因此可作为苯丙氨酸羟化酶的替代疗法。经过聚乙二醇修饰以后,半衰期延长,使用更加方便[23]。苯丙酮尿症是一种氨基酸代谢异常的常染色体遗传疾病,98% ~ 99%的患者是由苯丙氨酸羟化酶缺乏所致[24],在美国的患病率为1/15 000 ~ 1/10 000[25],约有3万名患者,是患者基数非常理想的罕见病。尽管有大量患者已经使用盐酸沙丙蝶呤(Kuvan)治疗,但Palynziq有疗效方面的优势,本品的年治疗成本高达20万美元,因此只要有3 000名患者选择这种疗法,其年销售额就有望突破6亿美元,科睿唯安对本品在2024年的销售额预测值为5.92亿美元。
15 Baricitinib
2018年5月31日,FDA批准了礼来的Janus激酶(JAK)1/2抑制剂baricitinib(商品名:Olumiant,9),用于肿瘤坏死因子(TNF)抑制剂应答不足的中重度类风湿性关节炎患者的治疗。之前其被FDA拒绝过1次[26],于2017年率先获得EMA的批准。FDA此次批准本品基于4项临床试验,其中2项(NCT01185353和NCT01469013 )是剂量相关性试验,结果显示:ACR20[美国风湿病学会改善标准,定义为肿胀及触痛关节个数(28个)有20%改善,以及患者对疼痛自我评价(VAS)、疾病总体状况的自我评价(VAS)、医生对疾病状况总体评分(VAS)、健康评估问卷(HAQ)和急性期反应物(ESR、CRP)等5项指标中,至少有3项改善20%,ACR50和ACR70以此类推]应答率随剂量的增加而提高。另外2项(NCT01721057 和NCT01721044)是为期24周的疗效确认试验,在传统缓解疾病的抗风湿性药物(cDMARDs)的背景治疗下,使用每日1次2 mg baricitinib或安慰剂治疗,24周后,治疗组的ACR20应答率分别为61%和45%,安慰剂组为42%和27%;ACR50应答率分别为41%和23%,而安慰剂组分别为21%和13%;ACR70应答率分别为25%和13%,而安慰剂组分别为8%和3%[27]。科睿唯安预测本品在2024年的销售额为12.02亿美元。
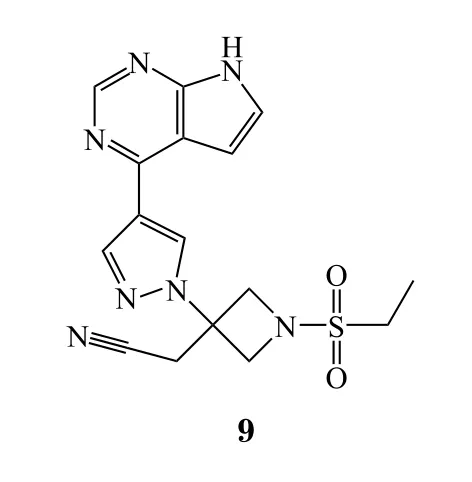
16 莫西菌素
2018年6月13日,FDA批准Medicines Development for Global Health的莫西菌素(moxidectin,10),用于盘尾丝虫病治疗。莫西菌素是一种大环内酯类驱虫抗生素,早在20世纪60年代问世,但美国一直未批准本品人用,因此莫西菌素也是典型的老药新批的品种。盘尾丝虫寄生于眼部或皮下组织,主要临床特征为眼部损害,可致失明,故又称河盲症。在一项有1 472名患者参与的临床试验(NCT 00790998)中,977名患者服用莫西菌素,475名患者服用伊维菌素,经过1个月的治疗,莫西菌素治疗组微丝蚴平均密度为每毫克皮肤0.1条,而伊维菌素治疗组为每毫克皮肤2.3条[28]。12个月后,莫西菌素治疗组微丝蚴平均密度为每毫克皮肤1.79条,而伊维菌素治疗组为每毫克皮肤9.83条。河盲症在发达国家已很少见,但在非洲却非常常见,本品的获批无疑是非洲人民的福音。
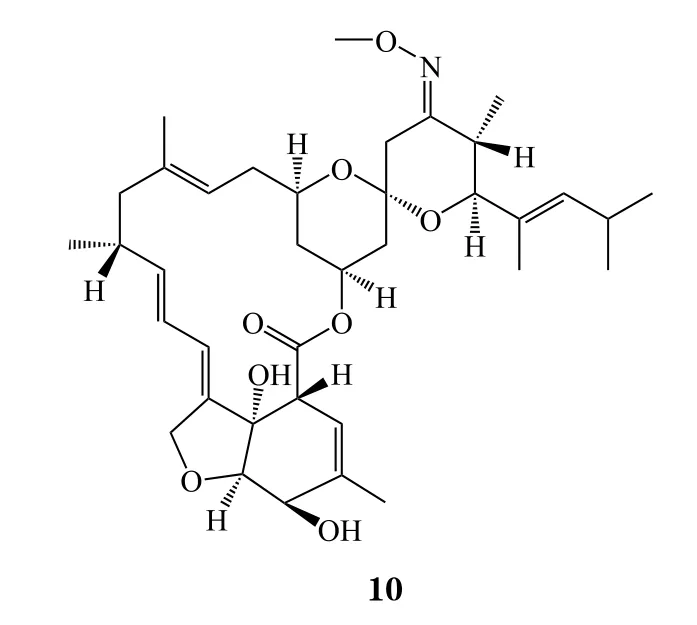
17 Plazomicin
2018年 6月25日,FDA批 准 了Achaogen公 司的新一代氨基糖苷类抗生素plazomicin(商品名:Zemdri,11),用于尿路感染治疗。在一项有609名成人患者参与的非劣效性临床试验(NCT02486627)中,本品治疗组的5 d复合治愈(临床意义上的治愈或改善、微生物根除)率为88%,而美罗培南治疗组为91.4%,达到统计学意义上的非劣效,治愈检验访视(test-of-cure visit,TOC)复合治愈(临床意义上的治愈、微生物根除)率分别为81.7%和70.1%,达到统计学意义上的优效[29]。自20世纪80年代以来,人类的感染疾病得到非常好的控制,虽然有部分新型抗生素被研发出来,但往往被限制使用,因此在过去30年里,制药巨头对抗生素的研发并不积极。随着近年来超级细菌的出现,新型抗生素的研发再次成为创新药的热门,因为本品对多重耐药性的格兰阴性菌有效,在2017年5月获得FDA突破性疗法认定,科睿唯安预测本品在2024年的销售额为0.78亿美元。

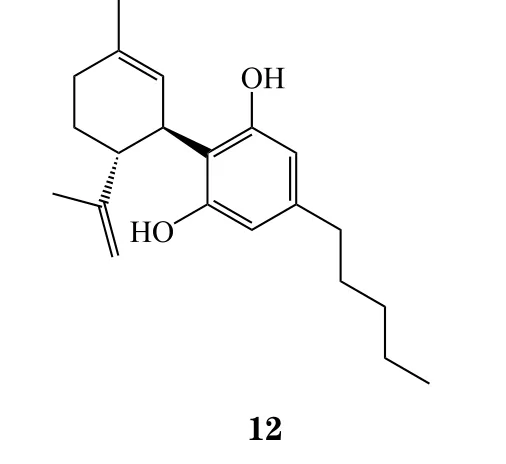
18 大麻二酚
在plazomicin获批的同一天,FDA还批准了GW公司的大麻二酚(cannabidiol,商品名:Epidiolex,12),用于Lennox-Gastaut综合征和Dravet综合征治疗。大麻二酚是从大麻中提取分离出的单体,是FDA批准的首个大麻类药物[30]。一项有225名Lennox-Gastaut综合征患者参与的多中心临床试验(NCT02224560)数据显示:大麻二酚治疗组患者的癫痫发作频率相比安慰剂组大幅下降,其中,10 mg · kg-1治疗组癫痫发作频率相比基线下降37.2%,20 mg · kg-1治疗组下降41.9%,而安慰剂组仅下降17.2%[31]。另一项国际多中心的临床试验(NCT02224690)也得到了相似的结果,86例Lennox-Gastaut综合征患者使用20 mg · kg-1的大麻二酚治疗,癫痫发作频率相比基线下降43.9%,而安慰剂组(n=85)仅为21.8%[32]。在治疗Dravet综合征方面,61例使用大麻二酚治疗的Dravet综合征患者,每月癫痫发作次数从12.4次下降到5.9次,而安慰剂组仅从14.9次下降到14.1次[33]。Lennox-Gastaut综合征和Dravet综合征都是罕见的癫痫综合征,治疗方案匮乏,本品疗效出色,而且是长期用药,非常有潜力成为重磅炸弹药物,科睿唯安预测本品2022年的销售额可达11.91亿美元。
19 Encorafenib
2018年6月27日,FDA批准了Array公司的encorafenib(商品名:Braftovi,13)与binimetinib(商品名:Mektovi,14)联合使用,治疗BRAFV600E或BRAFV600K阳性的转移性黑色素瘤。FDA批准本品是基于一项活性对照的三臂临床试验(NCT01909453)的结果,共计577名BRAFV600E或BRAFV600K阳性的转移性黑色素瘤患者参与了研究。患者按1 : 1 : 1比例分成3组,分别接受encorafenib与binimetinib联合治疗、encorafenib单药治疗或vemurafenib单药治疗。结果显示:encorafenib与binimetinib联合治疗组患者中位无进展生存期(PFS)达14.9个月,而vemurafenib单药治疗组仅为7.3个月[34]。

20 Binimetinib
在FDA批准Array公司的Braftovi同时,还批准了该公司的binimetinib(商品名:Mektovi),与encorafenib联合使用,治疗BRAFV600E或BRAFV600K阳性且无法手术的黑色素瘤。Binimetinib是一种有丝分裂原细胞外信号调节激酶(MEK)抑制剂,而encorafenib是一种以BRAFV600E基因为靶点的激酶抑制剂,FDA批准binimetinib上市,是基于二者的一项联合用药试验(NCT01909453)。在这种疗法之前,FDA已经批准葛兰素史克(GSK)/诺华的 trametinib + dabrafenib和罗氏的cobimetinib + vemurafenib用于BRAFV600E或BRAFV600K阳性的转移性黑色素瘤治疗,仅从PFS和OS数据对比而言,Array公司的encorafenib+binimetinib具有明显的优势(见表1),但GSK/诺华和罗氏的组合疗法都未达到重磅炸弹药物级别,Array的组合疗法要想成为重磅炸弹药物,依然有一定难度,科睿唯安对Array公司的组合疗法在2023年的预测销售额为5.55亿美元。
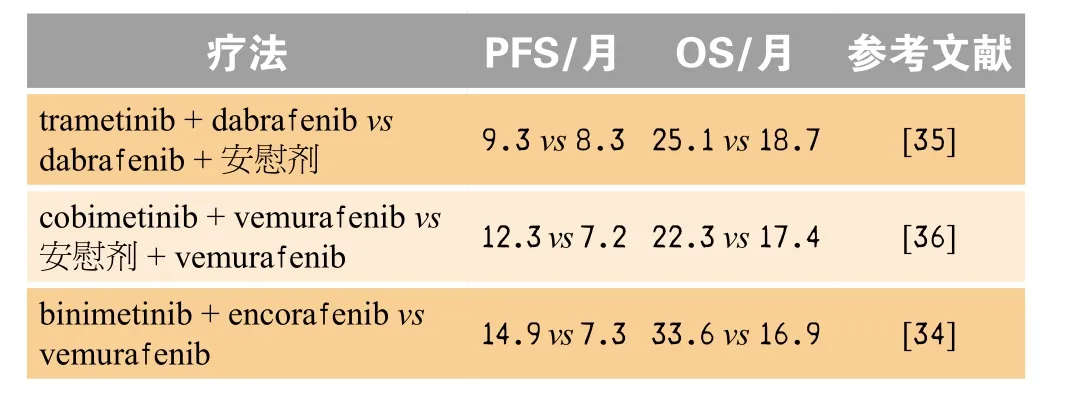
表1 BRAF V600E疗法对比Table 1 Effectiveness comparison of BRAF V600E treatment regimen
21 Tecovirimat
2018年7月13日,FDA批准了Siga公司的tecovirimat(商品名:Tpoxx,15),用于天花治疗。Tecovirimat是一种天花病毒p37蛋白抑制剂,FDA批准本品上市仅基于一项安全性试验和动物试验的结果。由于天花病几乎已经绝迹,为试药而在人身上接种天花病毒不符合伦理,因此并未开展tecovirimat的有效性临床试验,而是获准用新西兰兔替代,最终试验证实,tecovirimat治疗组相比安慰剂组生存率显著提高[37]。这是第1种获批用于对抗天花的药物,尽管世界卫生组织曾对外宣布,天花早在20世纪80年代已被根除,但长期以来人们一直担心天花被做成生化武器。FDA在公告中已经说明,批准本品是为了应对生物恐怖主义的威胁[38]。Siga公司也因tecovirimat获得了一张优先审评券,11月1日,Siga公司对外宣布以8 000万美元的价格向礼来转让其优先审评券[39],因此Siga公司可能“醉翁之意不在酒”,而在于优先审评券,这种创新药开发策略值得广大中小型企业学习和效仿。
22 Tafenoquine
2018年7月20日,FDA批准了GSK的tafenoquine(商品名:Krintafel,16),用于16岁及以上患者的间日疟根治(或复发预防)。FDA批准本品是基于2项临床试验的数据,试验一(NCT01376167)共有399名患者入组,其中260名患者接受本品联合氯喹治疗,139名患者接受氯喹加安慰剂治疗。经过6个月的治疗,tafenoquine加氯喹治疗组相比安慰剂加氯喹治疗组的复发率下降了76%。试验二(NCT01376167)是一项剂量相关性试验,结果与试验一类似。经过6个月的治疗,tafenoquine加氯喹治疗组84%的患者无复发,而安慰剂加氯喹治疗组仅为39%[40]。疟疾是一种寄生虫感染疾病,在发达国家几乎已经绝迹,因此本品对GSK而言,可能学术价值远大于经济价值,或许GSK也是醉翁之意不在酒,因为本品的获批,该公司获得一张宝贵的优先审评券。

23 Ivosidenib
在Krintafel 获批的同一天,Agios公司的新药——ivosidenib(商品名:Tibsovo,17)获FDA批准用于异柠檬酸脱氢酶-1(IDH1)基因突变的复发或难治性急性粒细胞白血病(AML)治疗。Ivosidenib是一种IDH1抑制剂,此次获批基于一项开放标签的单臂临床试验(NCT02074839)的结果,174名IDH1突变的复发或难治性AML患者参与了试验,中位随访时间为8.3个月,32.8%的患者获得完全缓解(无疾病迹象且血细胞计数完全恢复)或部分血液恢复的完全缓解(无疾病迹象且血细胞计数部分恢复)。AML是一种进展快速的血液肿瘤,据美国国家癌症研究所估计,2018年有19 520人被诊断为AML,其中10 670人将死于该病[41]。考虑到受众患者人群并不多,而且复发性或难治性AML已经有多个疗法获批上市,本品单靠AML适应证几乎不可能成为重磅炸弹药物,科睿唯安预测本品在2024年的销售额为6.21亿美元。
24 Elagolix sodium
2018年7月23日,FDA批准了艾伯维的促黄体激素释放激素受体抑制剂elagolix sodium(商品名:Orilissa,18),用于子宫内膜异位症的中重度疼痛治疗[42]。Elagolix的有效性已在2项共有1 686名患者参与的临床试验(EM-I 和 EM-II)中进行了评估,这2项试验设计几乎相同,患者被分为3组,分别每日1次口服本品150 mg,每日分2次服用本品200 mg或安慰剂。试验通过子宫内膜异位症每日疼痛影响量表(daily pain impact scale,DPIS)来评估药物的疗效,在EM-I试验中,经期疼痛评分相比基线下降超过0.81分或非经期盆腔疼痛评分相比基线下降超过0.36分,视为患者对药物应答。经过3个月的治疗,150 mg治疗组的痛经患者应答率为46%,非经期盆腔疼痛患者的应答率为50%,200 mg治疗组分别为76%和55%,安慰剂组则为20%和26%。在EM-II试验中,患者的应答标准有所提高,经期疼痛和非经期盆腔疼痛评分分别相较基线下降超过0.85分和0.43分,视为应答。3个月时,150 mg治疗组的痛经患者应答率为43%,非经期盆腔疼痛患者的应答率为50%,200 mg治疗组分别为72%和58%,安慰剂组则为23%和37%[43]。子宫内膜异位症是女性常见的疾病,影响了10%的生育年龄段女性,2项临床试验均说明elagolix剂量依赖性地降低了子宫内膜异位症患者的痛经或非经期盆腔疼痛。由于本品对骨密度影响较小,加之具有口服的优势,有望成为重磅炸弹药物,科睿唯安预测本品销售额将在2024年达到20.00亿美元。
(未完待续)
