一个中国X连锁肾上腺脑白质营养不良家系发生ABCD1基因突变(Ser342Ter)
2019-03-01胡琳晢南善姬隋冉冉卓明星
胡琳晢,南善姬,隋冉冉,卓明星,吴 杰
(吉林大学第二医院 神经内科,吉林 长春130041)
X连锁肾上腺脑白质营养不良(X-Linked adrenoleukodystrophy,X-ALD)是一种神经变性疾病,发病率大约为1∶15,000-30,000男性。该疾病的生化特征为饱和极长链脂肪酸(VLCFA)在脑白质、肾上腺和皮肤成纤维细胞中病理性地大量沉积。X-ALD的VLCFA的沉积与ABCD1基因突变有关[1]。我们现报道一个发生未发表的ABCD1基因突变(Ser342Ter)的中国X-ALD家系。
1 临床资料
先证者1:男,49岁,因记忆力减退6个月,加重2个月于2014年2月28日入院。患者入院前6个月无明显诱因逐渐出现记忆力下降,近2个月记忆力减退进行性加重,情绪容易激动,暴躁。既往外伤后右耳耳聋7年;吸烟史25年,30支/日,饮白酒6个月,饮酒量不详。入院查体:血压120/80 mmHg,神志清晰,言语流利,概测定向力、记忆力、计算力差,共济失调,余神经系统查体未见阳性体征。血清皮质醇测定:皮质醇5点-7点125.50 ng/ml(66-268);促肾上腺皮质激素 55.10 pg/mL (0.10-46);皮质醇15点-17点 77.06 ng/ml(22-154)。醛固酮测定(卧位) 63.80 pg/mI(59.5-173.9),醛固酮测定(立位) 90.21 pg/mI(65.2-295.7),余血液常规生化检查正常。心电图、胸片检查正常。头部MR:脑干、双侧小脑半球、双侧侧脑室后角旁可见斑片状长T1长T2信号(图1)。肾上腺CT:形态未见明显异常。临床诊断为肾上腺脑白质营养不良。住院期间,患者走路不稳、欣快、情绪容易激动,睡眠不良,便秘,4天后症状无明显改善出院。患者出院后症状进一步加重,脾气暴躁,开始酗酒。4月份,因精神症状,家人将其送至精神病院治疗数日。6月份,患者到北京协和医院检查代谢6项正常,极长链脂肪酸:二十二烷酸37.18 mg/l(11.52-28.18),二十四烷酸47.23 mg/l(10.68-28.72),二十六烷酸1.23 mg/l(0-0.5),C24/C22 1.27(0-1.24),C26/C22 0.03(0-0.02),确诊为肾上腺脑白质营养不良。10月份随访,患者痴呆、走路不稳,家人述曾走失1次,日常生活全部靠家人照顾。
先证者2,先证者1的弟弟,男,45岁,走路不稳、脾气暴躁1年于2014年9月28日到门诊就诊。自述9年前开始逐渐出现走路略不稳,但不影响日常活动。智能正常,能胜任会计师工作。查体:右侧跟膝胫试验略欠稳准,双下肢膝腱反射亢进。头部MRI:脑干、双侧小脑半球、双侧侧脑室后角旁可见斑片状长T1长T2信号。
随访该家系,共有3代人,13名成员。先证者的父亲健在,身体健康,母亲因脑血管病过世,生前无类似症状。除了2位先征者,其他兄弟姐妹、子女均无异常(图2)。
签署知情同意书后,采集先证者及其他家庭成员的外周血5 ml,进行ABCD1基因突变检测[2]。检测结果:先证者1(II3)1和先征者2(II5)ABCD1 外显子2,c.1025C>A,Ser342Ter,无义突变;ABCD1 外显子2,c.1047C>A,Val349Val,同义突变;ABCD1 外显子6,c.1548G>A,Leu516Leu,同义突变。其中,ABCD1基因外显子2,c.1025C>A(Ser342Ter)改变无报道,该位点改变导致编码氨基酸提前终止,提示为致病突变(如图3)。ABCD1 基因外显子2,c.1047C>A(Val349Val) 和外显子6,c.1548G>A(Leu516Leu)序列改变均为多态性位点。II4、III3、III5ABCD1 基因外显子2,c.1025C>A,Ser342Ter,杂合突变,为致病突变携带者。其他家庭成员ABCD1基因外显子2未发现序列异常。
2 讨论
我们对1个X-ALD家系进行ABCD1基因检测,发现一个目前为止尚未报道的突变c.1025C>A,Ser342Ter。该突变存在于第2个外显子,使第342个编码丝氨酸的密码子TCG改变为TAG(终止密码子),导致ABCD1基因提前终止翻译,形成异常ABCD1蛋白。自从X-ALD数据库建立,已经报道了超过1300种突变(http://www.x-ald.nl)。现在我们又增加了一种新的突变。
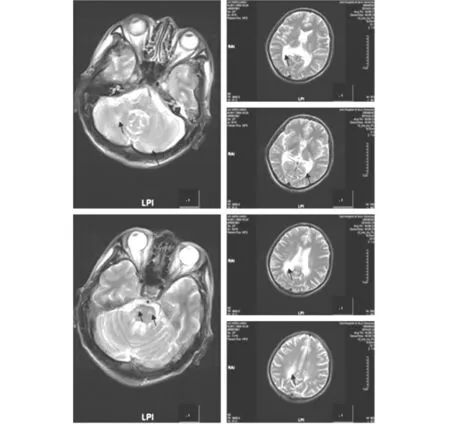
T2像示右侧小脑、双侧脑干腹侧,双侧侧脑室旁点片状高信号影

图2 X连锁肾上腺脑白质营养不良家系图

先证者1(II3)1和先征者2(II5)ABCD1 外显子2,c.1025C>A,Ser342Ter,无义突变。↓ 示C突变为A
X-ALD有很多临床表型,包括儿童脑型、青少年脑型、成人脑型、肾上腺脊髓神经病型(Adrenomyeloneuropathy,AMN)、Addison型、无症状型和杂合子型[2]。我们报道的这一家系中,先证者1发病半年后,病情迅速进展,符合脑型X-ALD(cerebral adrenoleukodystrophy,CALD)。 先证者2临床表现符合AMN型,虽然头部MRI显示脑部脱髓鞘改变,尚未发生急性进展性的脑白质脱髓鞘。此外,
该家系中还存在3个携带者,目前为止尚无异常。
综上,推测该家系的致病突变基因来自先证者的母亲,她极有可能是该突变的携带者。女性携带者发病很少见,2位先证者的女儿均为携带者,建议进行相关方面的遗传咨询。
