早发性家族性阿尔茨海默病家系的临床表型及基因突变分析
2019-03-01李燕坤庄顺芝陈礼锟张停亭崔理立李才明
李燕坤,庄顺芝,何 菲,陈礼锟,张停亭,崔理立,李才明
(1.广东医科大学,广东湛江524000;2.惠州市第一人民医院神经内科,广东惠州 516003;3.南方医科大学遗传学教研室,广东广州 510515)
阿尔茨海默病(Alzheimer′s disease,AD)是老年痴呆中最常见的一种[1],是以进行性认知功能障碍和记忆损害为特征的神经系统变性疾病[2]。人群发病率为1.0%~2.0%,并随着年龄增长而增高[3]。AD以65岁为界可分为早发性(early-onset AD,EOAD)和晚发性(late-onset AD,LOAD)2种,其中LOAD大约占94%。AD还可以依据是否与家族发病有关划分为家族性(familiar AD,FAD)和散发性(sporadic AD,SAD)[4]。在 LOAD 中以SAD的为主,在EOAD中则以FAD居多。目前已证实报道的β淀粉样前体蛋白(amyloid precursor protein,App)、早老素蛋白1(presenilin 1,PS1)、早老素蛋白2(presenilin 2,PS2)三个基因的突变是引起EOFAD的重要遗传致病因素[5-7]。目前我国有关EOFAD的遗传和临床资料报道不多。现我们发现一例EOFAD,并收集家系成员的临床资料及相关信息,对患者及其家系成员进行AD相关基因检测和比对,结合国内外相关文献,探讨基因突变引起该家系EOFAD的临床表型与遗传学特点。
1 材料与方法
1.1 临床资料收集
对该EOFAD家系先证者进行病史采集、神经系统体格检查、颅脑磁共振成像(magnetic resonance imaging,MRI)、脑电图、认知功能和精神状态检查,征得该患者与家系成员知情同意后,并将其与家系成员采血后送基因公司进行全外显子组基因检测;对12名家系成员进行病史采集、神经系统体格检查,TTC3基因检测。应用简易精神状态检查量表(mimi-mental state examination,MMSE)、蒙特利尔认知评估量表(Montreal Cognitive Assessment,MoCA)进 行 评 分 ,其 中MMSE评分标准为:总分0~30分,文盲组≤17分、小学组≤20分、中学以上≤24分则提示异常,MoCA评分标准:满分30分,≥26分属于正常。采用美国国立神经病、语言交流障碍和卒中研究所-老年性痴呆及相关疾病学会标准进行AD的诊断。
1.2 基因检测
采集Ⅰ1、Ⅰ2、Ⅱ2、Ⅱ3、Ⅱ6、Ⅱ7、Ⅱ8、Ⅱ9、Ⅲ2、Ⅲ3、Ⅲ4、Ⅲ5、Ⅲ6的外周血标本各约5 mL,放置于EDTA抗凝管中。其中Ⅰ1、Ⅰ2、Ⅱ3、Ⅱ7四人的血样送基因测序公司进行全外显子检测,确认可能引起EOFAD的基因突变位点。根据先证者的基因检测结果,对其余家系成员针对先证者的突变的基因进行检测。
2 结果
2.1 临床特征
该家系共有17名成员(图1),汉族,居住于广东省惠州市,其中男性9名,女性8名,其中一位成员于2017年3月因外伤意外死亡(Ⅱ5),家系中有4例AD患者(Ⅰ2、Ⅱ2、Ⅱ3、Ⅱ8),家系中AD患者符合AD相关疾病临床症状或体征。同时该家系符合EOFAD家系的临床诊断标准,属常染色体显性遗传。

图1 EOFAD家系谱图Fig.1 Pedigree of EOFAD
先证者(Ⅱ3)为男性,39岁,因“反应迟钝伴记忆力减退3年”于2016年10月9日步行入院后收治于本科。患者于3年前无明显诱因开始出现反应迟钝,伴记忆力减退(短期记忆能力下降),性格改变(孤僻),一般生活能自理,能自行走路。查体:意识清楚,对答欠切题,查体尚合作,理解力、定向力、判断力、计算力、记忆力均减退,心肺腹查体无特殊。双侧指鼻试验、快速轮替试验笨拙;双侧跟膝胫试验基本正常,闭目难立征可疑阳性,无神经系统局灶性缺损体征,MMSE得分19/30分(定向能力7/10分,语言功能7/8分,计算力1/5分,词语即刻回忆3/3分,延迟回忆1/3分,结构模仿0/1分),蒙特利尔认知评估评分18/30(视空间与执行能力2/5分,命名3/3分,注意5/6分,语言1/3分,抽象1/2分,延迟回忆2/5分,定向4/6分)(初中文化水平)。脑脊液检测及其余生化常规检查等均无明显异常。MRI结果示:幕上脑室系统普遍性轻度扩大,脑沟脑池增宽,脑实质内未见异常信号灶。MRI诊断为大脑弥漫性脑萎缩(图2A);MRS结果示:单体素,多脑叶多兴趣区采样示NAA峰较普遍性降低,但尚存在;部分Cho增高;其中左侧颞叶兴趣区NAA/Cr 1.34,Cho/Cr 1.13,NAA/Cho。幕上脑室系统扩大,脑沟脑池增宽。MRS结果提示神经元变性类疾病(图2B)。出院后规律服药:每晚多奈哌齐5 mg、奥氮平2.5 mg(因患者伴有性格改变,孤僻行为,不愿意与亲戚交往,惧怕他人伤害他)。2018年4月随访结果:患者曾走失2次,精神状态较前好转,能与人打招呼,但智力水平明显下降。
先证者之母(Ⅰ2),67岁,2年前外伤后发现“脑萎缩”,近两年发现智力下降,记忆力下降,偶尔交流不了,言语不流利,查体:行走缓慢,Rossolimo征阳性,双跟膝胫可疑阳性,心肺腹查体无明显异常。MMSE得分25/30分(定向能力9/10分,语言功能7/8分,计算力4/5分,词语即刻回忆3/3分,延迟回忆2/3分,结构模仿0/1分),蒙特利尔认知评估评分18/30分,(视空间与执行能力1/5分,命名3/3分,注意5/6分,语言1/3分,抽象2/2分,延迟回忆1/5分,定向5/6分)(初中文化水平)已确诊为AD,病情较重,目前需人照顾。

图2 家系成员的影像学检测Fig.2 Imaging examination results of family members
Ⅱ2,43岁,女性,30岁左右逐渐出现反应迟钝,记忆力下降。于2016年10月因偶发头晕半年就诊,偶发头痛,无其他特殊不适,前年在外院行头颅CT检查无异常,现入院后行头颅CT检查示双侧大小脑半球对称,脑干形态保持,脑实质未见明显异常密度灶。脑中线结构居中,脑室系统扩大,脑沟、裂、池增宽,颅板下未见异常密度影。CT诊断为脑萎缩(图2C)。蒙特利尔认知评估评分18/30分(视空间与执行能力1/5分,命名2/3分,注意4/6分,语言2/3分,抽象2/2分,延迟回忆2/5分,定向5/6分)(初中文化),已确诊为AD,出院后予规律服药(每晚多奈哌齐5 mg)。2018年4月随访:病情进展严重,记忆力明显下降,患者因经济关系未完善MMSE、MoCA、头颅MR或CT等检查。目前生活不能自理,需家属照顾。
Ⅱ5,37岁,女性,于2017年3月因外伤意外死亡,26岁时开始发病,去世前仅相当于5~6岁的智商,记忆力、计算力下降,言语表达不清,呆滞,走路不稳,大小便失禁,生活不能自理,查体:神智不清,言语含糊,右眼睑下垂,左眼球外展、内收障碍,瞳孔大小左侧:3 mm,右侧:2 mm。对光反射消失。双手轮转笨拙,左手明显,双上肢腱反射减退,双下肢腱反射消失,心肺腹查体无明显异常。MMSE得分16/30分(定向能力4/10分,语言功能8/8分,计算力1/5分,词语即刻回忆1/3分,延迟回忆1/3分,结构模仿1/1分),蒙特利尔认知评估评分12/30分(视空间与执行能力2/5分,命名2/3分,注意3/6分,语言0/3分,抽象0/2分,延迟回忆4/5分,定向1/6分)(高中文化),确诊为AD。
Ⅱ7,女性,35岁,目前暂为家系第Ⅱ代中正常人,无明显相关疾病表现,2016年头颅CT无异常,2018年复查头颅CT无异常,仍需继续跟踪随访。
Ⅱ8,男性,31岁,30岁时发病,逐渐出现反应迟钝,记忆力下降,暂无性格改变,生活能够自理,查体无明显异常,颅脑CT平扫示:双侧大脑半球、脑干及双侧小脑半球内未见明确异常密度影。中线结构居中,无明显移位;双侧侧脑室对称、稍扩大;脑沟脑裂稍增宽。颅骨未见异常征象。CT诊断为脑萎缩。MMSE得分20/30分(定向能力8/10分,语言功能6/8分,计算力2/5分,词语即刻回忆2/3分,延迟回忆2/3分,结构模仿0/1分),蒙特利尔认知评估评分20/30分(视空间与执行能力4/5分,命名2/3分,注意4/6分,语言2/3分,抽象1/2分,延迟回忆4/5分,定向3/6分),确诊为AD。

图3 家系成员TTC3基因的测序结果Fig.3 Sequencing results of the family member TTC3 gene
2.2 基因检测结果
将先证者Ⅱ3作为主患者与其父母作为对照,进行全外显子测序(图3)后发现:①先证者Ⅱ3在TTC3基因的第24个外显子发生c.2146C>T突变,导致所编码的TTC3蛋白的第716个氨基酸由原来的脯氨酸变为丝氨酸(p.Pro716Ser),该突变来源于自发变异。②先证者Ⅱ3在TTC3基因的第9个外显子发生c.758A>G突变,导致所编码的TTC3蛋白的第253位氨基酸由原来的酪氨酸变为半胱氨酸(p.Tyr253Cys),该突变来源于母亲。
将以上测序后的突变位点分别对家系中成员(Ⅱ2、Ⅱ6、Ⅱ7、Ⅱ8、Ⅱ9、Ⅲ2、Ⅲ3、Ⅲ4、Ⅲ5、Ⅲ6)进行检测后发现:(1)TTC3(c.2146C>T)突变组检测结果均无变异。(2)TTC3(c.758A>G)突变组仅在家系成员(Ⅱ2、Ⅱ7、Ⅲ2、Ⅲ3、Ⅲ4)检测出有杂合变异。
结果说明TTC3(c.2146C>T)突变组与EOFAD发病无明显相关性,而TTC3(c.758A>G)突变组在EOFAD发病可能存在一定相关,因其中Ⅲ2、Ⅲ3、Ⅲ4三个家庭成员年龄太小(<8岁),未达到该家系中患病年龄平均水平(35岁左右),但家系中有一例目前临床症状表现正常的成员(Ⅱ7)也存在有该位点突变,考虑其可能暂未发病,该基因突变基本符合家系共分离,呈常染色体显性遗传。
2.3 生物信息预测结果
利用PolyPhen-2对TTC3突变型蛋白进行有害性预测(图4),TTC3基因c.758A>G错义突变在PolyPhen-2软件上的预测是有害的,有害性评分是0.998(评分越接近1越有可能有害),且该突变点在人、小鼠、老鼠、狗、黑猩猩和长臂猿等物种中氨基酸序列分析显示高度保守。图5是该突变在各物种保守性分析预测结果。同样由SIFT软件预测结果显示,突变后的评分-3.902分(评分越低于-2.5分越有可能有害),认为该突变会影响表型(图6)。

图4 PolyPhen-2软件预测TTC3基因的突变对表型的影响Fig.4 PolyPhen-2 software predicts the effect of mutations in the TTC3 gene on phenotype
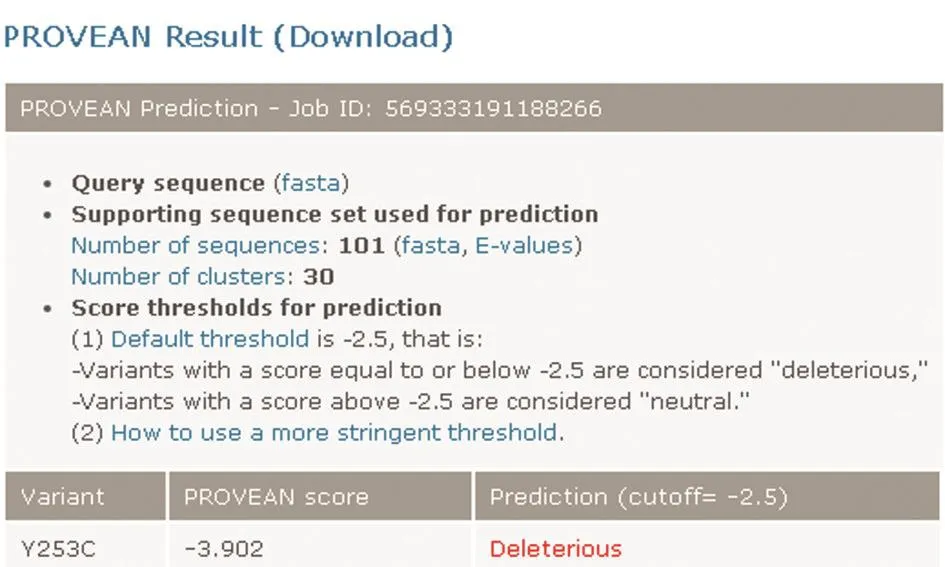
图6 SIFT软件预测突变对表型效应的影响Fig.6 SIFT software predicts the effect of mutations on phenotypic effects
3 讨论

图5 TTC3基因突变在各物种中保守性分析预测Fig.5 Conservative analysis and prediction of TTC3 gene mutation in various species
我们通过全外显子测序技术发现先证者在TTC3基因存在复合杂合突变:c.758A>G(p.Tyr253Cys)的错义突变,并且通过Sanger测序验证。c.758A>G(p.Tyr253Cys)为第9个外显子的编码区第758位碱基A突变为G,导致所编码的TTC3蛋白的第253位氨基酸由原来的酪氨酸变为半胱氨酸(p.Tyr253Cys),并且该点错义突变在家系中其他成员中满足家系共分离,且在生物信息学预测上为有害性,但此突变对蛋白复合物功能的实际影响需要进一步的动物实验及蛋白功能测定以证实。根据美国医学遗传学与基因组学学会联合美国分子病理学学会(ACMG-AMP)的序列变异分级标准指南,满足PM1+PP(1-5),属于可能致病性变异。
阿尔茨海默病是一种最常见的进行性神经系统退行性疾病,主要表现为认知功能。日常生活能力下降及精神行为异常等。以65岁为界,可以分为早发型阿尔茨海默病及晚发型阿尔茨海默病。其中约62%的早发型阿尔茨海默病有家族史,大部分以常染色体显性方式遗传。随着分子遗传学技术的进展,AD的遗传学研究取得了一定的进展。到目前为止,有数十种基因与AD发病相关。其中,EOFAD家系研究相关的基因主要与APP、PS1、PS2三个基因有关,其中最常见的为PS1突变引起。关于AD的基因突变分析多来自于国外,国内研究相对较少。
在本家系中,我们在先证者与其母亲和兄妹同患者中发现了TTC3基因的第9个外显子的第758位核苷酸序列发生A→G突变,该位置的氨基酸由原来的酪氨酸变成半胱氨酸,可能是导致早发性家族性阿尔茨海默病的发病,并且发病的家系成员的临床症状进展较快,且发病年龄较早(<30岁),首发症状为记忆力减退,反应迟钝,言语不流利,其中大部分患者最终均表现出典型的阿尔茨海默病的相关症状,如空间及时间定向力,计算、判断、语音能力减退,性格改变等,疾病进展较快,晚期智力和认知功能如同幼儿。其中有一名家系成员(Ⅱ7)也携带此突变基因,但未出现有相应的阿尔茨海默病的症状及影像学的改变,考虑到其家系中女性成员普遍发病年龄较男性晚,我们继续保持随访。此外,我们还发现有3名家系成员(第Ⅲ代)携带此突变,由于年龄较小(<15岁)以及目前尚无AD的相关表现,可能为症状前患者,我们将继续随访观察,以进一步证实基因检查的结果,同时也可以早预防、早治疗。
在过去研究当中,并未有报道过TTC3基因的调控作用与AD发病相关。对于TTC3基因的研究仅仅在唐氏综合征中,TTC3基因主要在21号染色体中唐氏综合征关键区域(DSCR)中,该基因是唐氏综合征候选基因之一[8]。TTC3蛋白是由2025个氨基酸组成的蛋白质,并且含有由富含半胱氨酸和组氨酸残基的短基序组成的典型H2-环指状结构域[9]。TTC3基因参与神经元增殖和分化,TTC3过表达能够对神经突触延伸起到强烈的抑制作用[10];TTC3表达分布主要位于神经元细胞中[11]。唐氏综合征(Down's syndrome,DS)病人常常表现出脑发育不正常,智力低下,年轻时发生阿尔茨海默病的发病率高[12]。DS中的脑形态特征在于皮质大小减小,小脑不成比例地变小,胆碱能神经元丢失[12]。神经元细胞系的功能研究表明TTC3在泛素化,Akt信号传导途径,负性细胞周期控制和神经元分化抑制中发挥重要作用[13-16]。因此,TTC3可能在多种AD发病相关途径中起作用。此外,这些过程在正常神经元和突触功能以及Aβ和tau蛋白病理学方面相互交织。同时TTC3蛋白是一种E3泛素连接酶,作为细胞存活的核心细胞内途径Akt信号传导的特异性末端调节剂[17]。Akt/mTOR(哺乳动物雷帕霉素靶标)信号传导是主要的细胞衰老途径,并涉及多种与年龄相关的人类疾病[18]。此外,大量证据表明Akt/mTOR信号作为关键因素参与在AD疾病中失调的神经元途径,损害TTC3蛋白功能的遗传变异可通过降低TTC3抵消活化的Akt的能力来促成AD易感性,从而促进Akt/mTOR 活性的增加[19],这可能与早发型AD的发病机制相关。
在先前报道的研究当中APP、PS1、PS2三个基因的突变是引起EOFAD的最常见的重要遗传致病因素,对此TTC3基因的突变在EOFAD发病当中所起的调控作用是一个新的发现,同时有可能作为一种新候选基因参与早发性家族性阿尔茨海默病的调控。此发现丰富了阿尔茨海默病家系资料库,同时为探讨早发型家族性阿尔茨海默病的发病机制提供了重要依据。
