作为Be2+磷光探针的含9-冠-3的铱配合物
2019-02-27胡媛媛姚永林周会东童碧海张千峰
潘 淼 胡媛媛 赵 卓 姚永林 周会东 童碧海 张千峰
(安徽工业大学冶金工程学院,分子工程与应用化学研究中心,冶金减排与资源综合利用教育部重点实验室,马鞍山 243002)
0 引 言
铍中毒是由铍及其化合物侵袭肺及其他器官引起的全身性疾病。国际肿瘤研究机构(IARC)的研究表明铍有致癌性[1-2]。但铍作为一种新兴材料在原子能、火箭、导弹、航空、宇宙航行以及冶金工业中是不可缺少的材料,应用广泛[3]。因此,通过高选择性和灵敏性的方法检测铍的含量非常重要。目前检测铍离子含量的常用方法有气相色谱法、离子色谱法、毛细管胶束电动色谱法、分光光度法、荧光光度法、原子吸收法、原子发射法、电感耦合等离子体质谱法、电感耦合等离子体原子发射光谱法和电化学法等[4]。但大多数方法都需要繁琐的前处理或操作过程,有些则需要贵重的仪器且选择性或灵敏度也不高。在此背景下,电致化学发光[5]、循环伏安法及电位计[6-10]等新方法被开发出来用于铍离子的检测。化学荧光探针由于高选择性和高灵敏度,易于检测等优点也被应用于铍离子的检测。例如用铍试剂Ⅱ实现了Be2+的比率计型荧光探针[11],用有机染料制备的双层纳米片实现了Be2+的可逆荧光探针[12]。但是由于铍的原子半径和离子半径特别小,电负性又相对较高,适合铍离子检测的荧光探针和方法非常有限。因此,寻找具有选择性高、响应快、检测方便的新型铍离子化学荧光探针仍然是一个挑战。
近年来,以磷光重金属配合物为化学传感器引起了人们的很大兴趣,这是因为磷光重金属配合物具有以下特点:较高的光热稳定性;发射波长不随所处环境的变化而变化;与有机荧光材料相比,具有较大的stokes位移和长的发射寿命,而长的发射寿命有利于使用时间分辨技术使磷光信号与背景的荧光信号相区分[13]。
9-冠-3是最小的冠醚分子,其冠醚空腔半径在0.16 nm左右[14]。而二价铍离子的半径非常小,为0.031 nm[11],甚至小于碱金属离子的半径,因此 9-冠-3是与二价铍离子最匹配的冠醚分子。因此,我们通过在环金属铱配合物的环金属化配体上引入9-冠-3铍离子接受器,合成了2种对铍离子有发光响应的环金属铱配合物,并研究了其物理化学性质和铍离子检测性能。
1 实验部分
1.1 试剂和仪器
试剂为市售分析纯产品,使用前经过标准方法处理干燥。核磁波谱由BRUKER AvanceⅢ400型超导核磁共振谱仪测试。高分辨质谱由AB Triple TOF 5600plus型质谱仪测试。紫外光谱由Shimadzu UV-3150型紫外可见分光光度计测试。荧光量子效率和寿命由Edinburgh(FLS-920型号)时间相关单光子记数荧光仪测试。荧光光谱由Shimadzu RF-5301PC型荧光分光光度计测试。氧化电位用循环伏安法测定,以二氯甲烷为溶剂,0.1 mol·L-1六氟磷酸四正丁基胺为支持电解质,二茂铁为外标,玻碳电极为工作电极,铂片为对电极,KCl饱和的银/氯化银为参比电极,用CHI60A型电化学工作站测定。
1.2 配体的合成
将 4′-醛基苯并-9-冠-3[15]0.500 g(2.4 mmol)溶于10.0 mL N,N-二甲基甲酰胺中,再滴入0.600 mL(5.6 mmol)2-氨基苯硫酚溶液,加入无水硫酸镁0.552 g(4.6 mmol),在氮气保护下反应24 h,反应完毕后,冷却至室温,用水和乙酸乙酯反萃取;将得到的有机相干燥,再用石油醚/乙酸乙酯体系硅胶柱层析,得到 4′-(2-苯并噻唑基)苯并-9-冠-3(BTZ9C3)的乳白色固体,产率40.8%。
1H NMR(400 MHz,CDCl3):δ8.03(d,J=8.7 Hz,1H,PhH),7.87(d,J=9.2 Hz,1H,PhH),7.77(s,1H,PhH),7.68(d,J=11.0 Hz,1H,PhH),7.41(d,J=46.9 Hz,2H,PhH),7.06(d,J=8.4 Hz,1H,PhH),4.52(d,J=6.7 Hz,2H,CH2),4.38(s,2H,CH2),3.99 ~3.86(m,4H,CH2)。MS-ESI:m/z=313.08(按[C17H15NO3S]+计算值:313.08)。
1.3 铱配合物的合成
1.3.1 配合物1的合成
将 0.182 g(0.51 mmol)IrCl3·3H2O、0.316 g(1.14 mmol)BTZ9C3、9 mL乙氧基乙醇和3 mL水放入35 mL圆底烧瓶中,溶剂脱气后用氮气保护,并在避光下于90℃加热24 h。冷却后过滤,沉淀用95%乙醇洗涤3次。真空干燥,得到深红色的铱二氯桥化合物0.360 g,产率为81.3%。
将70.0 mg(0.03 mmol)铱二氯桥化合物、26.8 mg(0.06 mmol)2,2′-bipyridine(bpy)溶于 5 mL,在室温下搅拌6 h;然后加入10倍物质的量(以铱二氯桥化合物的量为基准)的NH4PF6饱和甲醇溶液,在25℃下再搅拌6 h。减压下蒸除溶剂,过滤,沉淀用二氯甲烷/乙酸乙酯混合溶剂(1∶7,V/V)在硅胶上柱层析;收集橙红色部分,蒸除溶剂,得橙红色固体6.40 mg,即[Ir(BTZ9C3)2(bpy)]PF6(1),收率 22%。1H NMR(400 MHz,CDCl3):δ8.93 (s,2H,PyH),8.29(s,3H,PyH),8.15(d,J=1.8 Hz,1H,PyH),7.82(d,J=7.8 Hz,2H,PyH),7.72(s,1H,PhH),7.58(s,3H,PhH),7.56(d,J=8.4 Hz,4H,PhH),7.34(d,J=7.7 Hz,2H,PhH),7.15~6.97(m,2H,PhH),4.55(dd,J=8.0,4.9 Hz,1H,CH2),4.44(dd,J=7.8,5.3 Hz,3H,CH2),4.29(d,J=16.8 Hz,2H,CH2),4.26~4.12(m,2H,CH2),4.07~3.83(m,8H,CH2)。19F NMR(377 MHz,CDCl3):δ-72.23(s)。 HRMS-ESI:m/z=973.170 6(按[C44H36IrN4O6S2]+计算值:973.170 6)。
1.3.2 配合物2的合成
0.541 g(0.82 mmol)铱二氯桥化合物、0.218 g(1.02 mmol)3-三 氟 甲 基-5-(2′-吡 啶 基 )-1,2-二 唑(Hfppz)[21]溶于 25 mL 二氯甲烷中,在 25 ℃下搅拌 6 h。减压下蒸除溶剂,过滤,沉淀用二氯甲烷/乙酸乙酯混合溶剂(2:1,V/V)在硅胶上柱层析。收集橙红色部分,蒸除溶剂,得中性铱配合物[Ir(BTZ9C3)2(fppz)](2)的橙红色固体0.160 g,收率19%。1H NMR(400 MHz,CDCl3):δ9.27(t,J=9.0 Hz,1H,PyrazoleH),9.04(d,J=4.9 Hz,1H,PhH),8.38(d,J=8.9 Hz,4H,PhH),8.09(s,1H,PyH),8.01(d,J=6.6 Hz,1H,PhH),7.78(dd,J=18.4,6.8 Hz,3H,PyH),7.71 ~7.45(m,5H,PhH),7.11(d,J=30.8 Hz,1H,PyH),4.58(s,1H,CH2),4.49~4.27(m,1H,CH2),4.27~4.10(m,2H,CH2),3.89(s,1H,CH2),2.09(s,2H,CH2),1.70(s,4H,CH2),1.30(t,J=4.9 Hz,5H,CH2),0.92(s,1H,CH2)。19F NMR(377 MHz,CDCl3):δ-62.39(s)。 HRMS-ESI:m/z=1 029.145 4(按[C43H33F3IrN5O6S2]+计算值:1 029.145 4)。
1.4 离子检测实验
将配合物用乙腈和水(1∶1,V/V)溶液配制成10 μmol·L-1的溶液, 检测离子配制成 0.01 mol·L-1的水溶液进行测定。Be2+,Zn2+,Mn2+,Ag+,K+,Fe2+,Fe3+和Na+来自于硫酸盐,Cu2+,Cd2+,Mg2+,Ca2+,Cr3+,Co2+,Al3+和Ni2+来自于硝酸盐,而Li+和NH4+来自于氯化物盐。检测离子水溶液加入配合物溶液后摇匀,2 min后进行光谱测试。
1.5 配合物1的单晶结构测定
选取0.240 mm×0.220 mm×0.200 mm的单晶在Bruker公司的SMART APEXⅡ型X射线单晶衍射仪测定,采用经石墨单色器单色化的Mo Kα射线(λ=0.071 073 nm)作为衍射光源,于 296(2)K以 ω-2θ扫描方式收集数据。全部数据经Lp因子和经验吸收校正。晶体结构由直接法解出,部分非氢原子坐标在随后的数轮差值Fourier合成中陆续确定,理论加氢法给出氢原子在晶胞中的位置坐标。对氢原子和非氢原子分别采用各向同性和各向异性热参数进行全矩阵最小二乘法修正。全部结构分析计算工作采用SHELX-97程序系统完成[22]。
CCDC:1844315,1。

表1 配合物1单晶的基本参数和解析参数Table 1 Crystal data,data collection and structure refinement parameters for complex 1
2 结果与讨论
2.1 配合物的合成与表征
配体及配合物的合成路线如Scheme 1所示。首先,由4′-醛基苯并-9-冠-3和邻氨基苯硫酚脱水缩合以40%左右的产率得到配体4′-(2-苯并噻唑基)苯并-9-冠-3。然后该配体与三氯化铱进行反应,得到铱二氯桥中间体(dimer)[16],中间体粗产品不经提纯直接与不同的辅助配体反应得到目标产物。铱二氯桥与2,2′-联吡啶反应后,进行阴离子交换得到离子型铱配合物1;若铱二氯桥与Hfppz反应得到中性铱配合物2,两种产物的产率都在20%左右。产物的结构通过核磁共振氢谱和氟谱进行了确认。配合物的高分辨质谱中分子离子峰的质量与理论值吻合,进一步说明成功得到了目标产物。
Scheme 1 Synthetic route of iridium(Ⅲ)complexes

图1 配合物1的结构Fig.1 Structure of complex 1
为了进一步证明铱配合物的结构,配合物1用正己烷和二氯甲烷混合溶剂重结晶后,选取单晶测定了结构(图1)。从图中可以看出铱配合物中心形成了扭曲的八面体构型,铱离子与2个环金属化配体、1个bpy配体配位形成铱合物阳离子,平衡阴离子为PF6-。环金属化配体的N-N处于反式构型,而C-C处于顺式构型。铱离子与bpy配体的Ir-N键长分别为0.213 6和0.214 4 nm,大于铱原子与环金属化配体之间的Ir-N键长 (分别为0.206 1和0.206 6 nm),这主要是因为铱离子与bpy配体的Ir-N键与存在强供电子和反馈键相互作用的环金属化Ir-C键处于对位,受到了其影响[17]。Ir-C键的键长分别为0.200 7和0.202 6 nm,与文献报道的类似配合物的键长接近[18]。
2.2 配合物的光物理性能
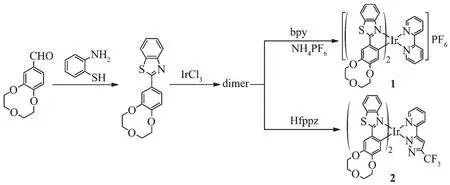
配合物1和2(10μmol·L-1)在二氯甲烷中的紫外-可见吸收光谱如图2所示。从图中可以看出配合物的光谱大体相似,这主要是由于它们的环金属化配体是相同的。350 nm以下的强烈吸收带(ε>1.6×104L·mol-1·cm-1)是典型的配体自旋允许的1ππ*跃迁。在350~460 nm附近的中等强度吸收带(ε>2.0×103L·mol-1·cm-1), 可以归属为金属到配体的单线态电荷转移(1MLCT)。而大于460 nm处的弱吸收带(ε<1.8×103L·mol-1·cm-1)可以归属为自旋禁止的三线态金属到配体的电荷转移(3MLCT)[19]。
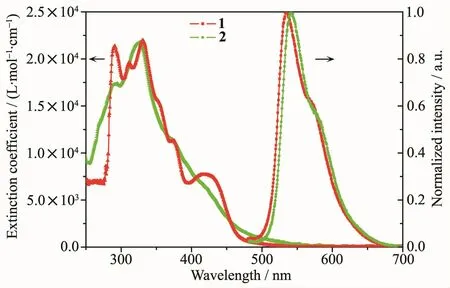
图2 铱配合物在二氯甲烷中的紫外光谱及发光光谱Fig.2 UV-Vis absorption and photoluminescence spectra of iridium(Ⅲ)complexes 1 and 2 in CH2Cl2
配合物1和2在二氯甲烷溶液中的光致发光光谱图也在图2给出。配合物1的最大发射波长为535 nm,是黄光发射,而配合物2的最大发射波长为541 nm,是黄绿光发射,发光相对于配合物1红移6 nm,这主要是由于配合物2的辅助配体的拉电子效应更强,使配合物的能隙增加,发光红移。由于磷光材料的发光受空气中氧气影响,因此将配合物以1%(w/w)的浓度掺杂到聚甲基丙烯酸甲酯(PMMA)中,制成薄膜以降低氧气的干扰,用来检测其发光量子效率和发光寿命。所得数据如表1所示,配合物1和2的发光寿命分别为3.01和2.58 μs,都在微秒级,是典型的磷光发射。同时,配合物1和2表现出较高的光致发光量子效率,分别为10.8%和45.0%,离子型铱配合物的发光量子效率较低。
2.3 配合物的电化学性能
为了研究配合物的能级水平,通过循环伏安法测定了它们的氧化还原电位。配合物1和2的氧化峰均为准可逆峰,为单电子氧化-还原过程,相对于二茂铁的氧化电位分别为1.48 V和1.21 V,换算成前线轨道HOMO能级分别为-5.60 eV和-5.35 eV,表明离子型铱配合物1的HOMO能级更低。

表2 配合物的荧光物理数据和电化学数据Table 2 Photoluminescence physical and electrochemical data of complexes 1 and 2
2.4 配合物的铍离子传感性能
为了研究配合物对铍离子的传感性能,首先进行了紫外-可见光谱检测实验,发现加入不同阳离子后配合物的紫外-可见光谱未有明显变化。发光光谱比一般的吸收光谱对于化学传感器和分析物之间的相互作用更敏感[18],因此继续进行发光光谱的检测实验。将2倍物质的量(相对于配合物)的各种阳离子加入到配合物溶液中后发现(图3a),配合物1在542 nm处的发射峰在Be2+存在下发光强度增加到原来的 126%,而其它离子(Li+,Na+,K+,Ca2+,Ba2+,Mg2+,Mn2+,Al3+,Ag+,Cu2+,Fe2+,Ni2+,Zn2+,Cr3+,Pb2+,NH4+和 Cd2+)引起的变化较小,相较于 Be2+变化不明显。同样,配合物2在546 nm的发射峰在Be2+存在下发光强度增加到原来的128%(图3b),而其它离子引起的发光强度变化小得多。这些现象表明配合物对Be2+有较强的选择性,有检测Be2+的能力。

图3 在2倍物质的量的不同金属离子存在下配合物1(a)和2(b)的发光光谱Fig.3 Luminescence emission spectra of complexes 1(a)and 2(b)in the presence of 2 equiv.of different metal ions
配合物对Be2+有选择性发光响应,为详细研究响应规律,进一步进行了配合物与Be2+的发射光谱滴定试验。从图4可以看出随着Be2+离子加入,2种配合物的发光都连续增强,直到加入2倍物质的量的Be2+后停止增强,表明配合物中每个冠醚基团都能与Be2+配位。增强过程中发光峰波长和发光光谱形状都保持不变。其中配合物1在541.5 nm处的发光强度增强到约 126%。 在 0~20 μmol·L-1的 Be2+浓度范围内发光强度呈线性响应,表明配合物1探针可以定量检测Be2+的浓度。线性方程为y=15.37+2.01x(R2=0.994),其中 y 是在给定的 Be2+浓度(μmol·L-1)时,541.5 nm处的发光强度,x是加入的Be2+浓度。计算得体系的检测极限(即相当于引起3倍空白标准偏差对应的浓度)为6.30μmol·L-1。与1类似,配合物2在546 nm处的发光强度增强到约128%,也可以定量检测 Be2+的浓度, 在 0~20 μmol·L-1的Be2+浓度范围内发光强度呈线性响应。线性方程为y=133.95+17.15x(R2=0.995),其中y为546 nm 处的发光强度,体系的检测极限为6.15μmol·L-1。以上结果表明,2种配合物都可作为Be2+的高灵敏度发光探针。此外,此滴定反应非常迅速,发光强度在3 min之内就不再变化,而且文献报道环金属铱(Ⅲ)配合物具低细胞毒性的特征,因此这种探针很可能适合在生物体内对 Be2+作实时的追踪[20]。
为了探索配合物作为的选择性化学传感器的实用性,做了一系列比较实验。先将2倍物质的量的干扰离子加入到配合物溶液中,然后再加入2倍物质的量的Be2+,实验结果如图5所示,图中黑柱代表加入干扰离子后的峰值光强,白柱代表继续加入Be2+后的峰值光强。当存在干扰离子时2种配合物的发光变化较小,在5%以内,继续加入Be2+后配合物1的峰值光强都增加,强度在原来的115%~124%之间,说明配合物1有较好的抗干扰能力。而配合物2的峰值光强也都增加,但在有Al3+存在时,只增加了6%;而其它离子存在时磷光的增加强度在原来的114%~131%之间。这可能是因为,虽然Al3+和Be2+两种离子的半径最为相近,分别为0.045和0.031 nm,但Al3+的电荷密度更高,不易与同样带正电荷的离子型配合物1接近,而易于与中性配合物2接近,从而使1和2表现出抗干扰能力的差别。
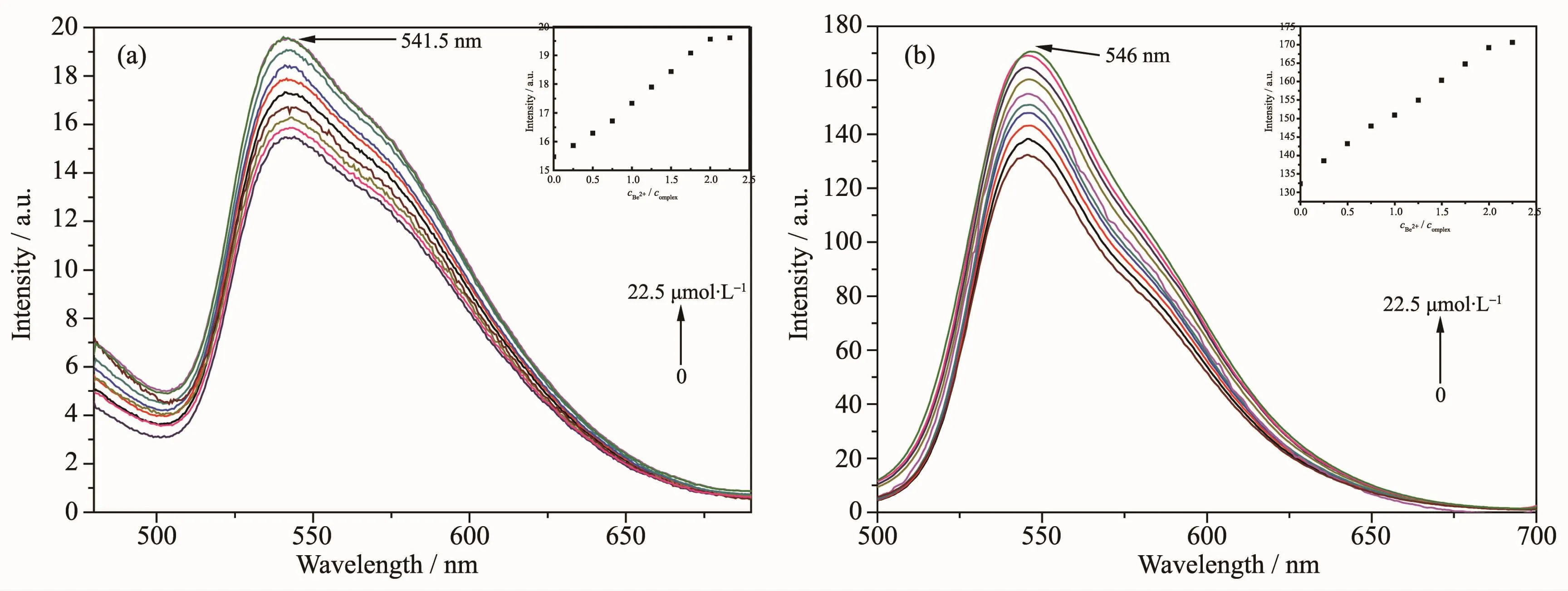
图4 在不同量的Be2+存在下配合物1(a)和2(b)的发光光谱变化Fig.4 Changes in the emission spectra of complexes 1(a)and 2(b)with various amounts of Be2+
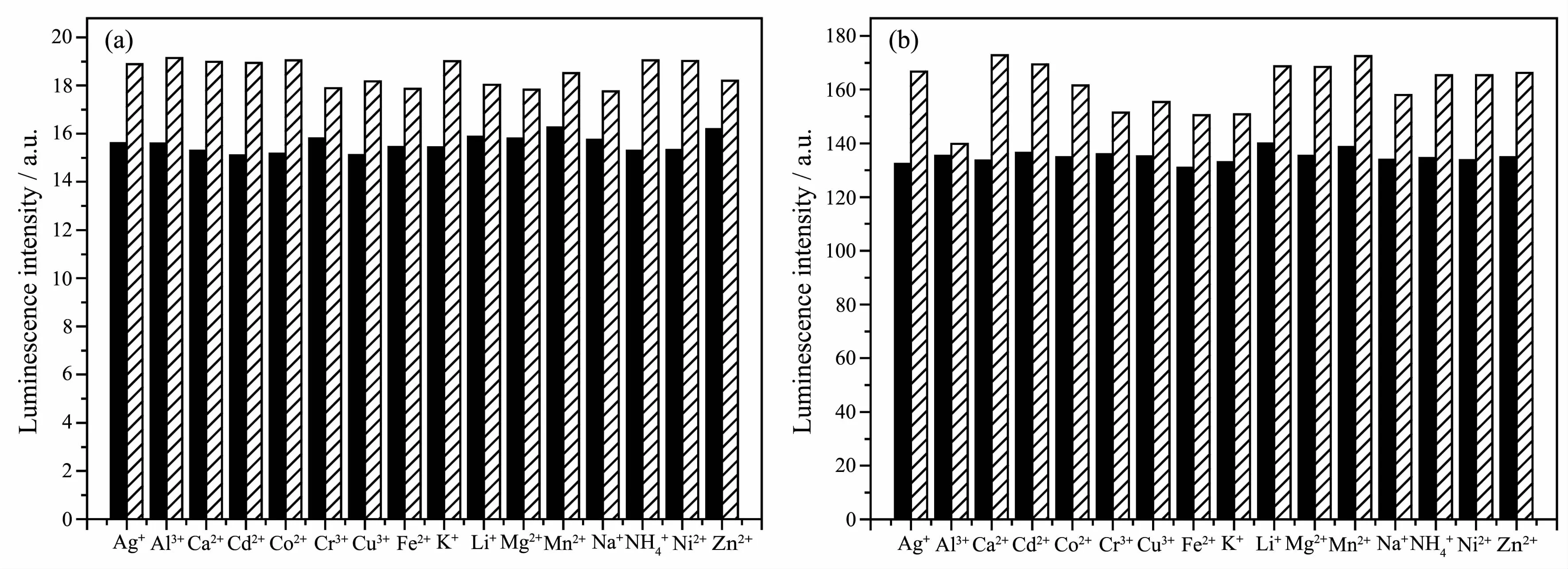
图5 在乙腈水溶液中1(a)和2(b)对各种金属离子的发光响应Fig.5 Luminescence responses of complexes 1 and 2 to various metal ions
对于Be2+使配合物发光增强的原因,我们推断,由于Be2+的离子半径只有0.031 nm,是所有金属离子中是最小的,而9-冠醚-3也是所有冠醚中最小的冠醚,因此两者尺寸最为匹配,能发生较弱的配位反应。配合物上冠醚基团在与Be2+配位后刚性增强,非辐射跃迁降低,导致发光增强。
3 结 论
合成了2种含有9-冠-3基团的铱配合物,并研究了其光物理性能,结果表明2种配合物为黄绿光发光,有较高的发光量子效率。2种配合物对Be2+都有发光增强的选择性识别效果,化学计量比为1∶2,最低检测限低至6.0μmol·L-1。抗干扰能力方面,离子型配合物1的抗干扰能力较好,而中性配合物2受Al3+的干扰较大。实验结果表明结合9-冠-3基团对Be2+的选择性和铱(Ⅲ)配合物高效发光的优点,可以实现新型高选择性的磷光传感器,为新型磷光传感器提供了思路。该工作将有利于实现低浓度Be2+的高选择性快速检测,减少Be2+对环境的污染。
