ATP7A基因内含子突变致Menkes病1家系2例报告
2019-01-25张晓青孙素真吴文娟
张晓青 孙素真 吴文娟 刘 康 王 晶
1 病例资料
先证者:男,4个月8 d,因“间断抽搐1 d”于2016年3月26日入河北省儿童医院(我院)。1 d前无明显诱因出现抽搐,表现为双眼向一侧斜视,一侧肢体阵挛性抽动,每次持续半小时至数小时缓解,缓解后如常,不伴肢体活动障碍,不伴发热、咳嗽,无呕吐、腹泻。发作间期意识清楚。自发病以来精神、吃奶欠佳,体重增长缓慢,大小便正常。
系G2P2,足月剖宫产娩出。生后2个月能追光追物。逐渐发现患儿运动发育落后,至就诊时仍不会抬头,肢体活动少,逗笑少。其哥哥因出生时缺氧于当地医院诊断“脑性瘫痪”。父母非近亲婚配,无类似疾病家族史。
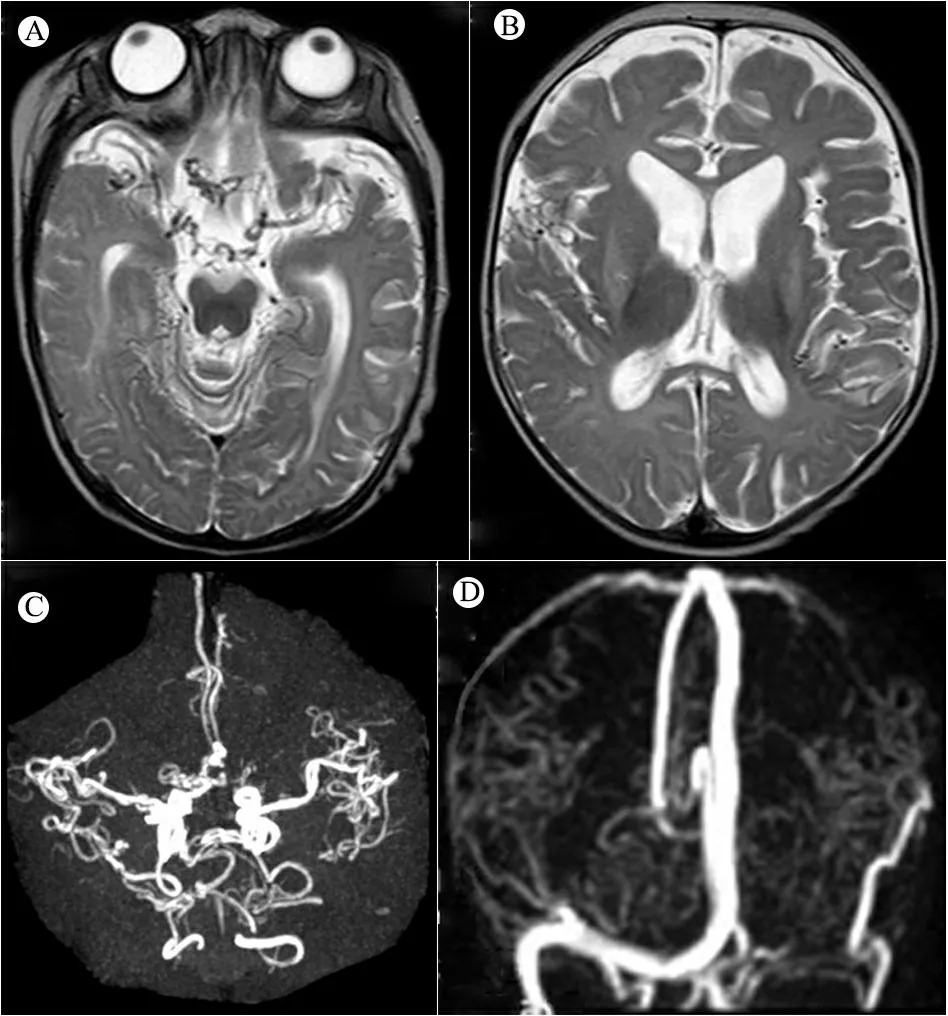
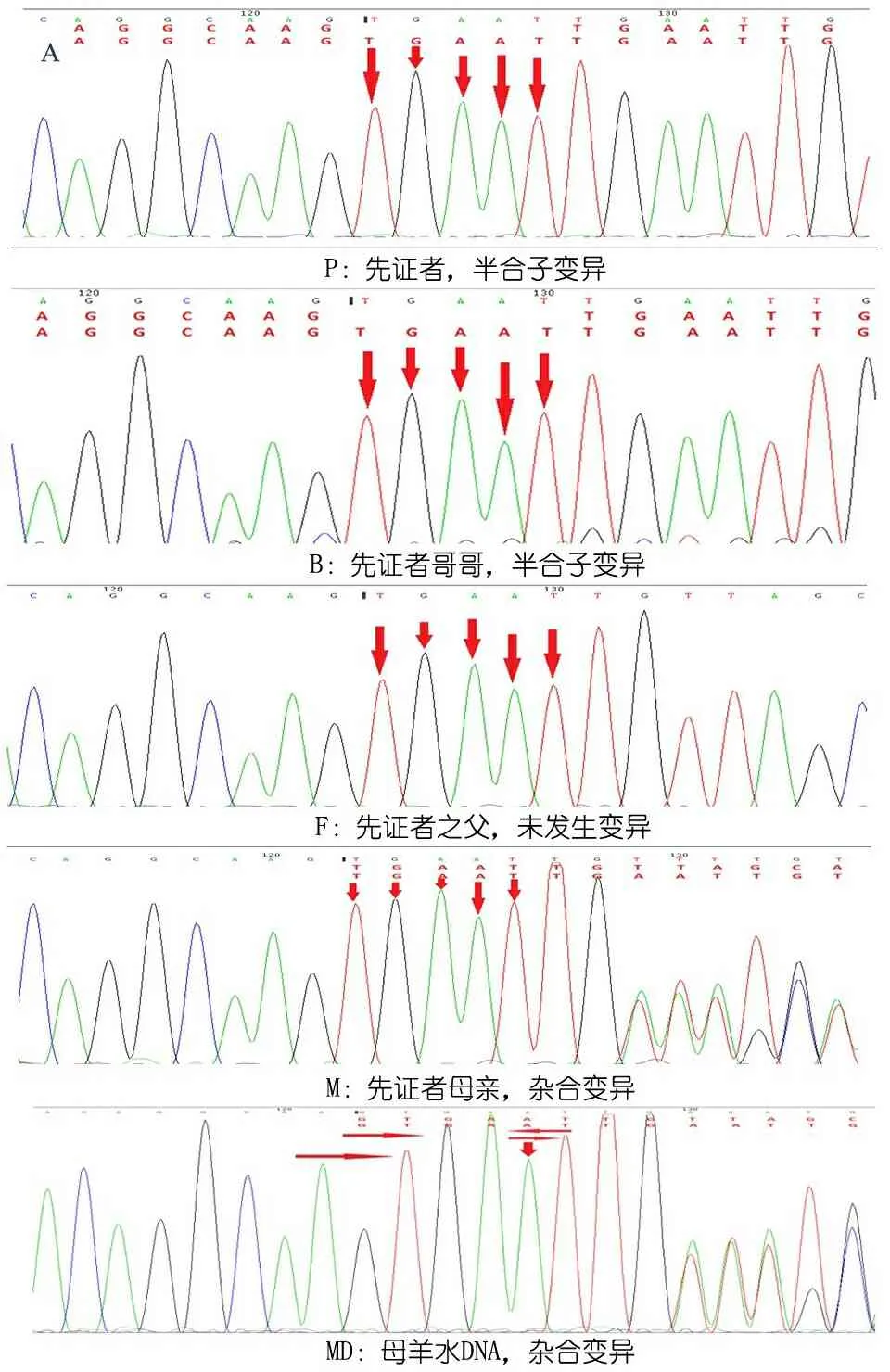
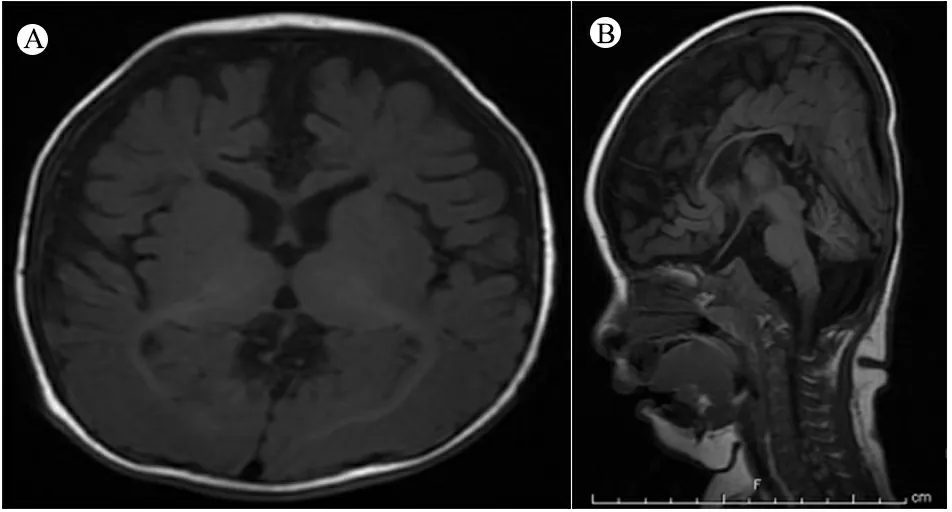
查体:体重5.5 kg( 图1例1面容及头发 注 A~B:患儿4月8天;C~E:患儿2岁9月 图2病例1患儿颅脑MRI及血管图像 注 A:T2WI:外侧裂、鞍上池、环池多发迂曲血管影,脑沟增宽;B:T2WI:基底节区对称片状长T2信号,侧脑室大,脑外间隙增宽,脑沟增宽;C:MRA:颅内大血管迂曲走形,远端分支血管扭曲成团;D:MRV:双侧大脑表浅静脉增多、成团扭曲。静脉窦未见明显异常。 例2 :先证者哥哥,足月顺产,出生体重2 660 g,出生时有羊水Ⅲ°污染,生后Apgar评分1 min 9分、5 min 10分。生后1 d因“吃奶差、反应差”就诊于当地医院,查体发现患儿营养差,皮下脂肪薄头发干燥、发黄。诊断“新生儿感染、新生儿代谢性酸中毒、先天性遗传代谢病?”,予对症治疗好转出院。7月龄时因“智力运动发育落后”就诊于我院,查体:皮肤松弛,毛发色浅,注视、追物不灵活,声音刺激治疗2年,智力运动发育无明显改善。与先证者一起行基因检测,提示存在与先证者相同的基因突变(图3),查铜蓝蛋白(北京大学第一医院)0.09 mg·L-1。 图3ATP7A基因测序图 注ATP7A基因c.2172+5_2172+6insTGAAT突变家系验证,P:先证者,B:先证者哥哥,F:先证者之父,M:先证者之母,MD:先证者之母再孕羊水DNA;红色箭头表示突变位置 不敏感,不会抬头、翻身、独坐,无主动抓物及眼神交流,四肢肌力减低,肌张力高,腱反射活跃,双巴氏征阳性。查血代谢筛查、染色体核型分析未见异常。颅脑MRI(图4)示,双侧大脑半球白质对称性减少,胼胝体体部变薄,脑室、脑沟、脑池增宽加深,考虑脑白质发育不良。按“脑性瘫痪” 图4例2患儿颅脑MRI 注 T1WI横轴位显示,双额颞脑外间隙、前纵裂增宽,脑沟增宽,双侧内囊前肢信号减低,髓鞘化延迟;B:T1WI矢状位显示,胼胝体薄,考虑脑发育不良 患儿的母亲再孕,于妊娠20周时取羊水细胞行胎儿ATP7A基因突变分析,进行产前诊断。提示患儿编码区第 2172 号编码区内含子c.2172+5_2172+6insTGAAT表型正常的携带者(图3MD),胎儿出生后发育正常。 本文2例患儿的ATP7A基因突变为内含子突变(c.2172+5_2172+6 insTGAAT),在万方、中国知网、PubMed和EBSCO数据库中检索类似病例,检索时间为建库至2018年12月1日。中文数据库以“万方医学网”为例,检索式为((Menkes病) ANDATP7A) AND 内含子突变。英文数据库以PubMed为例,检索式为("menkes kinky hair syndrome"[MeSH Terms] OR ("menkes"[All Fields] AND "kinky"[All Fields] AND "hair"[All Fields] AND "syndrome"[All Fields]) OR "menkes kinky hair syndrome"[All Fields] OR ("menkes"[All Fields] AND "disease"[All Fields]) OR "menkes disease"[All Fields]) AND (("introns"[MeSH Terms] OR "introns"[All Fields] OR "intronic"[All Fields]) AND ("mutation"[MeSH Terms] OR "mutation"[All Fields]))。 检索并筛选到2篇与ATP7A内含子突变有关的报告,均为英文文献[1,2],共报告4例患儿,其中2例OHS和1例经典Menkes病的ATP7A mRNA中含有新的假外显子,假外显子插入在外显子10和11、外显子16和17、外显子14和15之间;另1例OHS的表型是由1个剪接位点突变引起的,该突变涉及内含子6在铜结合域的+6位置,称轻度OHS表型(OMIM 30001.0006)。 Menkes病,即卷毛病,是一种罕见的先天性铜代谢异常引起的进行性神经性退行性疾病,尤以中枢神经系统和结缔组织损害为主,患儿多在3岁前死亡[3]。Menkes病属于X-连锁隐性遗传病,由ATP7A基因突变引起P 型铜转运 ATP 酶缺乏导致。国外报道发病率为1/300 000~1/360 000[4]。大多数患儿为男性,但也有些女性患者的报道[5-11]。 ATP7A基因定位于Xq12-q13.3,转录的23个外显子为编码铜转运的p型ATP酶。截至 2017年12月31日,人类基因突变数据库共收录ATP7A基因的变异317 种,其中一些与疾病无关[16],大约有1/3的情况下出现新生突变,但突变类型与临床病程无明显相关性[17],到目前为止国内共有与“Menkes病”相关的文献50篇,行ATP7A基因检测的13篇,均表现为经典型Menkes病,未发现ATP7A基因内含子突变所引起的Menkes病。国外文献中关于ATP7A基因深层内含子突变的报道仅2篇,4例患儿中,2例OHS和1例经典Menkes病患儿的ATP7A mRNA中含有新的假外显子,另1例OHS是由一个剪接位点突变引起的,该突变涉及内含子6在铜结合域中的位置+6[1,2]。在所有报道病例中内含子突变的存在增加了隐性供体剪接位点的一致值(consensus value),导致强剪接位点,所有病例一致值增加超过10%。一致值高于80的剪接位点是强剪接位点,而一致值在65~70的剪接位点是弱剪接位点,因为只有少数是激活位点,一致值的相对变为10%或更多可能影响剪接功能。这些激活的供体剪接位点都与高分受体位点结合使用。因此,这些突变似乎是通过激活新的供体剪接位点来激活位于内含子中的伪外显子,插入到内含子之间的伪外显子导致了异常氨基酸过早终止密码子,这些转录本都不能编码功能蛋白,可能通过无义介导的衰变(NMD)机制降解。编码蛋白都缺乏大部分C末端部分,使其不太可能具有任何残余活性。只有存在于患者体内的少量野生型ATP7A转录本才有望编码功能蛋白,存在野生型转录本可以解释轻度OHS表型。在未受影响的个体中发现,与经典Menkes病相比,只有2%~5%的正确拼接的mRNA水平就足以发展为OHS,而所报道的内含子突变经典型Menkes病仅表达0.2%的野生型转录本。 本文2例患儿符合经典型Menkes病诊断,基因检测未发现ATP7A基因存在大片段变异,而有ATP7Ac.2172+5_2172TGAAG(编码区第2172号核苷酸后内含子中第5与第6位核苷酸间插入TGAAT)的剪切变异,母亲为表型正常的携带者,符合Menkes病的遗传方式。但由于技术限制,未测定患儿体内野生型转录本的表达比例,但结合颅脑MRI及实验室检查提示铜蓝蛋白明显降低,支持诊断经典型Menkes病。该内含子突变位点此前未尚无报道,丰富了Menkes病的基因谱,为进一步研究内含子突变对疾病的影响提供了临床依据。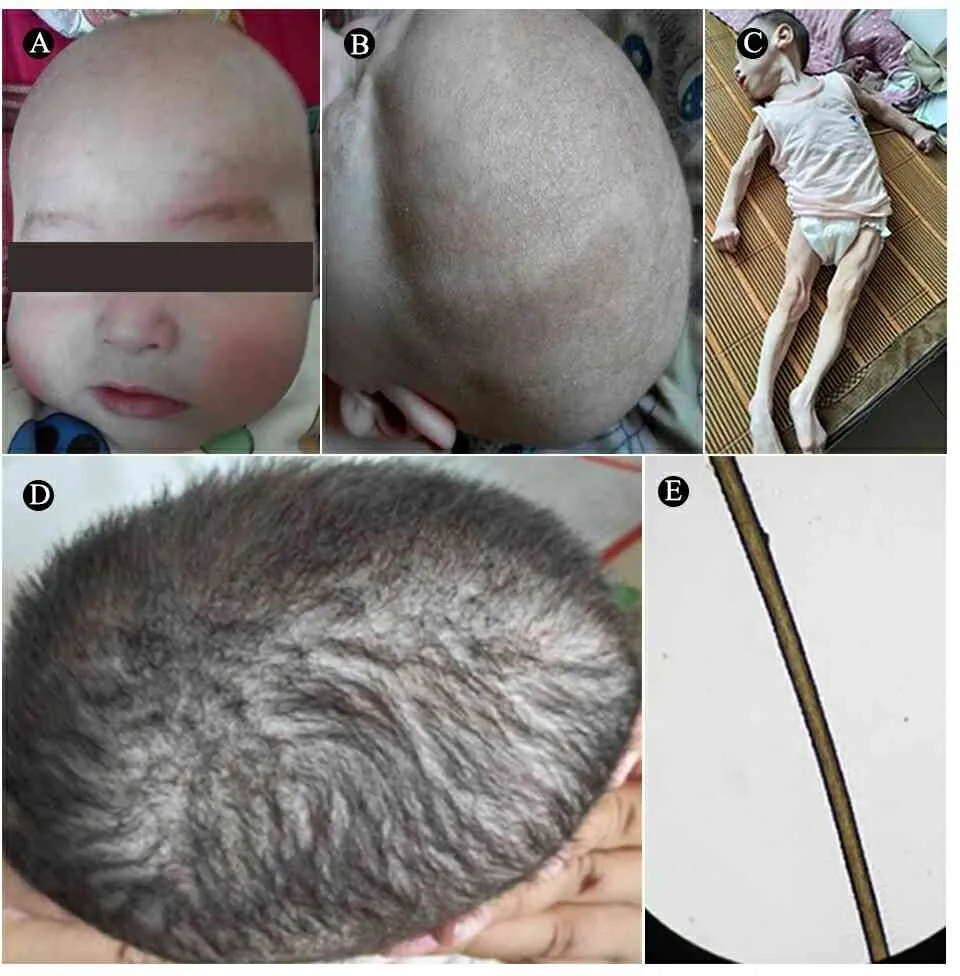

2 文献复习
3 讨论
