Cr掺杂Cu2O的光催化性质的第一性原理研究
2019-01-22吕巧雅李龙龙李亚方毛金花赵燕杰刘立强李鲁艳
吕巧雅,李龙龙,李亚方,毛金花,陈 婷,赵燕杰,刘立强,李鲁艳,*
( 1.山东建筑大学 理学院,山东 济南 250101;2.山东建筑大学 材料科学与工程学院,山东 济南 250101)
近年来,无机半导体材料的光催化技术在环境治理和能源转化等领域显示出广阔的应用前景[1-5]。TiO2和ZnO目前是最常用的光催化剂,但两者的禁带宽度在3.00-3.40 eV,只能在仅占太阳光5%的紫外光照射下激发[6]。而立方结构的Cu2O是一种禁带宽度为2.20 eV的金属缺位P型半导体,它对氧的吸附能力强、吸收系数高、无毒、制备成本低,并且在占太阳光45%的可见光照射下能进行光催化反应,所以在光催化、半导体技术、太阳能转换、磁存储设备、生物传感、防腐涂料和农药制备等方面起到很大作用[7-13]。
图1是常见半导体光催化剂的带边位置以及带隙宽度图[14-17]。从图1中可以看出,Cu2O的导带比能进行还原反应的能量更负,价带比能进行光催化氧化反应的能量更正,所以Cu2O非常适合作为可见光反应的催化剂,但是本征Cu2O在可见光区的光催化效率比较低。近年来,研究者发现在半导体结构中进行掺杂是提高半导体光催化效率的有效途径。Vaiano等[18]发现,Pr掺杂可以减小ZnO的光学带隙,使ZnO在可见光区域存在光催化活性;Jiang等[19]发现,Au掺杂可以提高Cu2O在可见光区域的光催化活性;Zhang等[20]发现,Zn掺杂可以使Cu2O在可见光区域的光催化活性增强;彭健等[21]发现,随着Cl掺杂浓度的增大,Cu2O的禁带宽度逐渐减小,Cu2O对可见光的吸收能力得到明显改善;截至目前对Cr掺杂Cu2O的研究报道极少。为了更好地探究Cr掺杂对Cu2O的电子结构和光催化活性的影响,本研究采用第一性原理的方法计算并分析了体系的形成能、电子结构和光学性质。

图 1 常见半导体光催化剂的带边位置以及带隙宽度[14-17]
1 计算方法和模型
1.1 计算方法
本研究采用基于密度泛函理论结合赝势平面波方法的Castep模块对Cu2O的122超晶胞结构进行结构优化和计算[22,23]。电子和电子之间的交换关联势使用广义梯度近似(GGA)和Perdew-Burke-Ernzerhof(PBE)方法[24],平面波的截止能量Ecut选取为480 eV,赝势采用超软赝势,用422的k点网格对简约第一布里渊区进行积分取样,采用Broyden-Fletcher-Goldfarb-Shanno(BFGS)法则对超晶胞进行了驰豫[25],系统总能收敛达到210-6eV/atom、每个原子上的力低于0.5 eV/nm时驰豫停止。经上述参数优化后,Cu2O晶胞的晶格常数为0.4286 nm,其基本上与实验得到的数值(0.4275 nm)是一致的[26]。对于电子结构及光学性质的计算,k点网格设置为633,总能的收敛精度达到10-6eV/atom。
1.2 理论模型
为了进一步推动Cu2O在可见光区中的应用,提高其催化效率,为实验提供理论指导,计算并分析了四种体系的能量、电子结构和光学性质。图2(a)是本征Cu2O结构;图2(b)是用一个Cr原子替换Cu2O中的一个Cu原子, 形成掺杂浓度是4.17%的Cr原子掺杂体系,写作Cu2O-1Cr;图2(c)是用两个Cr原子分别替换距离近的两个Cu原子,形成掺杂浓度是8.33%的近邻Cr原子掺杂体系,写作Cu2O-2Cr-near;图2(d)是用两个Cr原子分别替换距离远的两个Cu原子,形成形成掺杂浓度是8.33%的远邻Cr原子掺杂体系,写作Cu2O-2Cr-far。

图 2 Cu2O、Cu2O-1Cr、Cu2O-2Cr-near和Cu2O-2Cr-far的122超晶胞结构,红色球为O原子,蓝色球为Cu原子,黄色球为Cr原子
2 结果与讨论
2.1 形成能分析
为了表征掺杂体系的稳定性,计算了缺陷的形成能,具体见公式(1)。
Eform=Etotal-ECu2O+nECu-nECr
(1)
式中,Etotal是掺杂体系的总能量,ECu2O是本征Cu2O的能量,ECu和ECr分别是单个Cu原子和单个Cr原子的能量,n指被替代的Cu原子的数量,其中,Cu原子和Cr原子的能量按照最稳定的单质计算。经计算,Cu2O-1Cr、Cu2O-2Cr-far和Cu2O-2Cr-near三种掺杂体系的结合能分别约为-0.07、-0.06和-0.10 eV,它们的形成能均小于0,表明Cr容易掺杂进Cu2O晶体内,这三种掺杂体系是稳定的。
2.2 电子结构分析
为了研究体系的电子结构,分别计算了体系的电子能带、态密度和分波态密度。为了和掺杂体系进行比较,首先计算了如图3(a)所示的本征Cu2O的能带图,从图3(a)可以看出,计算得到的Cu2O是禁带宽度为0.52 eV的直接带隙半导体材料,与已有文献报道的带隙值非常接近[19,27],而明显小于实验值(2.20 eV),这主要是密度泛函对交换关联能考虑不足所引起的共性问题[28]。为此我们进一步使用GGA+U方法对Cu2O体系进行了计算,结果表明,引入在位库仑能(U)除了使Cu2O体系的带隙得到一定修正外,所得的结论与GGA方法得到的基本一致。并且已有文献显示采用GGA方法计算Cu2O掺杂体系能够给出比较可靠的结果[19,27,29],因此,接下来的讨论均是基于GGA方法得到的结果展开的。

图 3 本征Cu2O的能带图、总态密度图和分波态密度图
图3(b)-3(e)是体系的总态密度图和分波态密度图,从总态密度图3(b)发现,Cu2O的价带部分可以分为(-7.50)-(-4.50) eV能量的下价带区域和(-4.50)-0 eV的上价带区域。由分波态密度图3(b)-图3 (e)发现,Cu2O上价带主要由Cu 3d电子态主要贡献,下价带主要由O 2p态主要贡献,导带部分主要由Cu 3p和Cu 4s电子态主要贡献。
研究了一个Cr掺杂,即掺杂浓度是4.17%时体系的电子结构,计算得到的总态密度图和分波态密度图见图4。由图4可知,由于Cr原子的掺杂,体系显示金属性质,价带向下移动,价带顶到费米能级有0.67 eV的能量差,这个数值要比计算得到的本征Cu2O的带隙大。在Cr掺杂的Cu2O体系中,Cr 3d和Cu 3d轨道的相互排斥作用使体系的价带下移,所以价带中的电子想要跃迁到非占据态需要更大的能量,吸收边将向高能端移动,光学带隙发生蓝移。从分波态密度图4(b)-4(e)可以看出,杂质带Cr 3d态主要位于带隙中,跨越了费米能级,(-0.67)-0 eV能量被电子完全占据,0-0.68 eV能量在费米能级以上,没有被电子占据。另外,带隙中很少部分的电子态由Cu 3p和Cu 4s态贡献。

图 4 Cu2O-1Cr的总态密度图和分波态密度图
图5为两个Cr原子8.33%掺杂时远邻构型的总态密度图和分波态密度图。由图5(a)可知,体系显示金属特性,价带下移,从分波态密度图5(e)中可知,由于远邻Cr原子的存在,带隙中的电子态仍然主要由Cr 3d态贡献。与一个Cr原子4.17%掺杂的计算结果相比较,价带顶到费米能级的能量差增大到0.85 eV,所以,随着Cr掺杂浓度的增加,吸收边将继续向高能端移动,光学带隙的蓝移现象增强;同时Cr 3d态的峰值增强且局域性减弱,这主要是由于随着Cr掺杂浓度的增大而引入了更多的3d电子态引起的。另外,带隙中Cu 3p态、Cu 4s态和O 2p态贡献有所增强。
为了研究掺杂距离对Cu2O电子结构的影响,计算了两个Cr原子8.33%掺杂时近邻构型的总态密度图和分波态密度图,具体见图6。由图6可知,和两个Cr原子远邻掺杂体系相同,体系显示金属性质,带隙中存在的杂质态主要由Cr 3d态贡献,很少部分是由Cu 3p态、Cu 4s态和O 2p态贡献。进一步比较图5(e)和图6(e)发现,近邻远邻两种掺杂体系的Cr 3d态的电子占据情况和局域性有所不同,说明在相同Cr掺杂浓度下,不同的原子构型对材料的物理性质是有影响的。

图 5 Cu2O-2Cr-far的总态密度图和分波态密度图

图 6 Cu2O-2Cr-near的总态密度图和分波态密度图
2.3 光学性质分析
根据量子力学的观点,可以使用表面电子态的时间相关扰动来描述系统中光子与电子的相互作用。占据和未占据状态之间的转换是由光子吸收或发射引起的,激发引起的光谱可以被认为是导带和价带之间的联合态密度。采用第一性原理研究材料的光学性能主要是通过介电函数得出的,介电函数的实部(ε1)和虚部(ε2)可以由如下公式定义[30,31]:
(2)
(3)

(4)
2.3.1介电函数虚部分析
图7为四种体系的介电函数虚部图。由图7可知,本征Cu2O在可见光能量范围内电子跃迁很弱,分别在3.40和7.70 eV处存在两个主峰,结合态密度图3(b)-3(e)分析可知,在3.40 eV处的主峰应该是电子由Cu 3d态向导带跃迁即上价带向导带跃迁引起的;7.70 eV处的峰应该主要是电子由O 2p态向Cu 3d态跃迁导致的。
对于Cu2O-1Cr体系,其低能端的介电函数虚部明显比本征Cu2O体系要大,说明掺杂体系的电子可以通过更低的能量发生跃迁。通过分析态密度图4,带隙中存在被电子部分占据的Cr 3d电子态,电子可以由费米能级以下的占据态跃迁到费米能级以上的非占据态,从而发生带内跃迁,这种带内跃迁所需的能量比本征Cu2O价带导带间电子跃迁所需的能量要小,更容易产生电子空穴对,所以低能量范围内的电子跃迁主要由Cr 3d态电子的带内跃迁引起的。
与Cu2O-1Cr体系相比,Cu2O-2Cr-far体系在低能端(0.20 eV附近)有一个更强的峰,结合态密度图4和图5分析可知,Cu2O-2Cr-far带隙中跨越费米能级的Cr 3d态比Cu2O-1Cr的明显增强,说明有更多的电子可以通过小的能量发生带内跃迁,所以0.20 eV处的峰也主要是由Cr 3d态中电子的带内跃迁导致的。Cu2O-2Cr-near体系在0.20 eV处同样存在一个峰值,但是要比Cu2O-2Cr-far体系的峰值明显增强,说明与Cu2O-2Cr-far体系相比,Cu2O-2Cr-near体系会有更多的电子可以通过很小的能量发生带内跃迁。
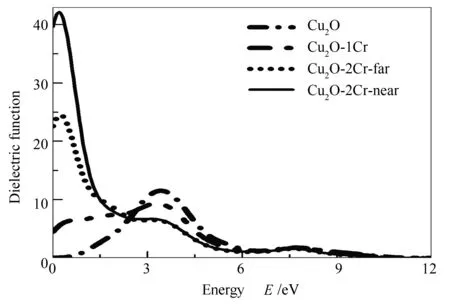
图 7 四种结构的介电函数虚部图
2.3.2吸收光谱分析
为了进一步分析本征Cu2O、Cu2O-1Cr、Cu2O-2Cr-far和Cu2O-2Cr-near四种体系的光催化性能,计算并分析了体系的吸收光谱,具体见图8,这里采用剪刀差的方法处理了吸收谱,以便与实验的吸收边相吻合[32]。由图8可知,四种体系的吸收范围均很宽,吸收部分都主要位于紫外光区域,并且在高能部分的振幅几乎相同,这与介电函数虚部图7中的高能端处的函数值几乎相吻合,说明不同掺杂浓度和结构构型主要影响材料在长波长段的物理性质,而对短波长段的性质影响很小。
从图8中还可以看出,本征Cu2O在可见光区域有很少的吸收,与图7中本征Cu2O在可见光区域电子跃迁很弱相对应。而Cu2O-1Cr、Cu2O-2Cr-far和Cu2O-2Cr-near三种体系在可见光范围内的吸收峰均有不同程度的增强,可见光范围内的吸收强度与介电函数虚部在低能量处峰的强度是对应的,增强的光吸收主要来源于掺杂引入的Cr 3d杂质态,而带隙变化对其影响很小。
进一步分析还可以发现,在可见光区域,Cu2O-2Cr-near是三种掺杂体系中吸收最强的,Cu2O-2Cr-far次之,Cu2O-1Cr最弱,说明Cr掺杂可以提高Cu2O在可见光区域的吸收强度,随着Cr掺杂浓度的增大,Cu2O在可见光区域的吸收强度增强;并且Cr原子近邻掺杂比远邻掺杂更能提高Cu2O在可见光区域的吸收强度。

图 8 四种体系的吸收光谱图,蓝色区域表示可见光能量范围
3 结 论
基于第一性原理分别计算并分析了本征Cu2O、Cu2O-1Cr、Cu2O-2Cr-far和Cu2O-2Cr-near四种体系的缺陷形成能、电子结构和光学性质。结果表明,本征Cu2O是直接带隙半导体,并且在可见光区域的吸收强度很低;三种掺杂体系结构稳定,显示金属特性,在可见光区的吸收均比本征Cu2O强,且Cr的浓度是8.33%的近邻掺杂最能提高Cu2O在可见光区域的吸收强度;四种体系在高能端的振幅相同,说明Cr掺杂对材料在短波长段的性质影响很小。基于以上研究,Cr掺杂确实可以提高Cu2O在可见光区域的光催化效率,为Cu2O在光催化方面的发展提供实验参考和理论基础。
