淡红侧耳子实体多糖的分离纯化及结构探析
2019-01-14田有秋贾金霞束旭任晓婕关月谢红许杨洁王昱沣
田有秋,贾金霞,束旭,任晓婕,关月,谢红,许杨洁,王昱沣
(南京农业大学 食品科学技术学院,江苏 南京,210095)
淡红侧耳是一种侧耳科、侧耳属的食用菌[1],其色鲜味美,蛋白质、粗纤维、维生素和氨基酸等含量丰富[2],作为一种兼食用、药用及观赏的食用菌而逐步被人们接受。多糖是组成生物体的一种高分子碳水化合物,不仅是细胞的结构材料,而且为细胞活动提供能量;它在细胞分化、代谢、生长、胚胎发育、免疫应答、抗血栓和抗肿瘤等方面发挥着重大作用[3]。淡红侧耳子实体粗多糖具有抗氧化等活性[4],是一种重要的活性成分。针对淡红侧耳的研究目前主要包括营养成分分析[5-6]、提取条件优化[7]、深层发酵[8]、和菌丝体多糖[9]等方面,对其子实体多糖的纯化、纯化后的多糖结构和理化性质的研究尚未见报道。多糖结构与其生物活性息息相关,对纯化多糖的结构进行研究不仅有助于进一步开展药理学、毒理学研究以及结构修饰改造等,更可以为多糖类新药的开发提供理论依据。
本研究以实验室前期对该多糖的提取研究结果为基础[10],首次对所获得的淡红侧耳粗多糖进行分离纯化,并对其纯化组分进行分子质量测定及单糖组成分析,采用紫外光谱、红外光谱及刚果红实验对其结构进行初步鉴定,且对其理化性质进行研究,为淡红侧耳多糖的进一步研究和应用提供依据。
1 材料与方法
1.1 材料与试剂
淡红侧耳,由广东银新现代农业股份有限公司提供。
DEAE-纤维素-52,葡聚糖标准品,Sigma公司;Sephadex G-150,台州市路桥四甲生化塑料厂;单糖标准品,上海源叶生物科技有限公司;甲醇:(色谱纯),KBr:(光谱纯),无水乙醇,国药集团化学试剂有限公司;乙腈(色谱纯),苯酚、刚果红、葡萄糖糖醛酸、K2SO4、H2SO4、NaCl(分析纯),南京化学试剂股份有限公司;咔唑、三氟乙酸、1-苯基-3-甲基-5-吡唑啉酮(PMP),阿拉丁试剂。
1.2 仪器与设备
层析柱,上海煊盛生化科技有限公司;STARTER 3100冷冻干燥机,奥豪斯仪器有限公司;LNG-T83B 离心浓缩仪,太仓华利达实验室设备公司;EU-2600R紫外可见分光光度计,上海昂拉仪器有限公司;HL-2S恒流泵,上海沪西分析仪器有限公司;Nicolet IR200 红外光谱仪,美国Nicolet公司;Waters 1525高效液相色谱仪,美国Waters公司;DBS-100电脑全自动部分收集器,上海沪西分析仪器有限公司;LC-20AD岛津高效液相色谱仪,美国岛津公司; RE52-99旋转蒸发仪,上海亚荣生化仪器厂;
1.3 方法
1.3.1 淡红侧耳粗多糖的提取
将淡红侧耳子实体干品洗净、烘干后粉碎,称取2.0 g细粉,按固液比1∶30.6 (g∶mL),提取温度95 ℃,提取时间3.9 h的条件进行水提醇沉,抽滤后得粗多糖粉末;将粗多糖粉末复溶至1.0 mg/mL,采用大孔阴离子交换树脂D315按树脂用量0.1 g/mL,pH 6,45 ℃,120 min的条件进行静态脱色;采用Sevage法对脱色后的多糖溶液进行脱蛋白;透析48 h后冷冻干燥,得淡红侧耳粗多糖[10]。
1.3.2 淡红侧耳粗多糖的分离纯化
1.3.2.1 DEAE-纤维素-52 阴离子层析柱纯化淡红侧耳多糖
称取80.0 mg淡红侧耳粗多糖溶于8 mL去离子水中,上样于层析柱(Φ26.0 mm×300.0 mm),装柱体积1 BV (设定1 BV=133 mL);分别用0、0.1、0.3、0.5、0.7 mol/L NaCl溶液进行梯度洗脱(流速1 mL/min,10 min/管);苯酚-硫酸法[11]检测偶数管多糖含量,绘制洗脱曲线,收集不同组分;50 ℃浓缩至约30 mL,去离子水透析48 h,将透析液真空冷冻干燥,得多糖各组分[12]。
1.3.2.2 Sephadex G-150层析柱纯化淡红侧耳多糖
分别称取20.0 mg上述多糖组分溶于5 mL去离子水中,上样于层析柱(Φ16.0 mm×300.0 mm),装柱体积1 BV (设定1 BV=50 mL);用去离子水洗脱(流速0.3 mL/min,10 min /管);以管数为横坐标,A490为纵坐标,绘制洗脱曲线,根据峰值分别收集不同组分;50 ℃浓缩至约30 mL,去离子水透析48 h,冷冻干燥后得多糖终纯化产物。
1.3.3 淡红侧耳多糖的纯度及相对分子质量测定
1.3.3.1 高效液相色谱条件
Waters 1525 高效液相色谱仪(配2410示差折光检测器和Empower工作站);检测柱为UltrahydrogelTMLinear (Φ7.8 mm×300.0 mm);流动相为0.1 mol/L NaNO3;柱温30 ℃,流速0.9 mL/min,进样量20 μL。
1.3.3.2 标准曲线的绘制
称取不同相对分子质量的葡聚糖标品50.0 mg于25 mL容量瓶中,用流动相溶解,定容;上样前用0.45 μm滤膜过滤,以保留时间为横坐标,相对分子质量的对数值为纵坐标制作标曲。
1.3.3.3 样品的测定
将淡红侧耳纯化多糖用流动相溶解,同上操作,将保留时间代入标曲,计算相对分子质量。
1.3.4 淡红侧耳多糖的单糖组成分析
1.3.4.1 高效液相色谱条件
LC-20AD岛津高效液相色谱仪(配SPD-20A紫外-可见检测器和LC solution色谱数据工作站);色谱柱:Thermo C18色谱柱(Φ4.6 mm×250.0 mm,5 μm);流动相:0.1 mol/L PBS (pH 6.7)和乙腈(体积比为83∶17);柱温30 ℃,流速0.7 mL/min,进样量20 μL;检测波长245 nm。
1.3.4.2 混合单糖标品衍生化
分别称取10种单糖标准品各3.0 mg,溶解到1 mL去离子水中;分别取0、20、40、60、80、100 μL混合单糖标品溶液,补水至100 μL,再加入100 μL NaOH溶液(0.6 mol/L),混匀;取100 μL混合液,加入等体积的PMP-甲醇溶液(0.5 mol/L),混匀,70 ℃水浴100 min,30 min后加入100 μL HCl (0.3 mol/L)中和,于50 ℃浓缩仪中旋干溶液;依次加入去离子水、氯仿各1 mL,混匀静置分层后吸取水相,如此重复3次,最终水相用0.45 μm水系滤膜过滤后进行高效液相色谱分析[13-14]。
1.3.4.3 样品的测定
分别称取3.0 mg PDP-1、PDP-2-1和PDP-2-2,用去离子水配成3 mg/mL溶液;取100 μL多糖溶液,加100 μL TFA (4 mol/L),于120 °C干燥箱中反应2 h;冷却后加入200 μL甲醇,于50 ℃浓缩仪中旋干,如此重复3次后,分别加100 μL去离子水进行衍生化,0.45 μm水系滤膜过滤后进行高效液相色谱分析。
1.3.5 理化性质的测定
1.3.5.1 总糖含量
分别取0、0.2、0.4、0.6、0.8、1.0、1.2、1.4 mL葡萄糖标准液(40 μg/mL)和1 mL多糖样品溶液(0.05 mg/mL),用去离子水补至2 mL;加入1 mL苯酚溶液(质量分数为6%),摇匀后迅速加入5 mL浓硫酸,混匀,静置20 min后,测A490。以葡萄糖质量为横坐标,A490为纵坐标,绘制标准曲线。将多糖样品测得的吸光值带入标曲,进而求得多糖样品中的总糖含量[11]。
1.3.5.2 蛋白质含量
分别取0、0.2、0.4、0.6、0.8、1.0 mL牛血清白蛋白标准液(100 μg/mL)和1 mL多糖样品溶液(3 mg/mL),用去离子水补至1 mL;加入5 mL考马斯亮蓝G-250,摇匀后测A595。以牛血清白蛋白质量为横坐标,A595为纵坐标,绘制标准曲线。测得样品吸光值,依据标曲计算蛋白质含量[15]。
1.3.5.3 糖醛酸含量
分别取0、0.05、0.10、0.15、0.20、0.25、0.30、0.35 mL葡萄糖醛酸标准液(200 μg/mL)和1 mL多糖样品溶液(0.14 mg/mL),用去离子水补至1 mL,加入5 mL四硼酸钠-硫酸溶液(9.54 mg/mL),混匀,沸水浴10 min,冷却后加入0.2 mL咔唑-乙醇溶液(1.25 mg/mL),混匀,沸水浴15 min,冷却后测A530。依据标准曲线计算多糖样品中的糖醛酸含量[16]。
1.3.5.4 硫酸基含量
分别取0、0.10、0.12、0.14、0.16、0.18、0.20 mL硫酸钾标准液(0.6 mg/mL),用1 mol/L盐酸补至0.2 mL,加入3.8 mL TCA(30 g/L);加1 mL氯化钡-明胶溶液(5 g/L)混匀,记为A1,加1 mL明胶溶液(5 g/L),记为A2;混匀后静置20 min,测A360。取10.0 mg多糖样品,溶于1 mL盐酸(1 mol/L),于100 ℃水解6 h,冷却,50 ℃浓缩至干;加1 mL去离子水复溶,取0.2 mL复溶液同上处理。以硫酸钾质量为横坐标,A1、A2差值为纵坐标,绘制标准曲线。依据标曲计算多糖样品的硫酸基含量[17]。
1.3.6 淡红侧耳多糖的光谱分析
1.3.6.1 紫外光谱分析
分别将淡红侧耳粗多糖和纯化多糖样品配制成0.5 mg/mL的溶液,于200~400 nm进行紫外-可见光谱扫描,观察其在260~280 nm有无吸收峰[18]。
1.3.6.2 红外光谱分析
取适量KBr于100 ℃干燥箱中烘干至恒重,称取100.0 mg干燥KBr,于玛瑙研钵中研磨至细,用压片机压成透明薄片后用于空白实验基线校正;再分别称取1.0 mg多糖样品与100.0 mg干燥KBr,充分研磨后压成透明薄片,于4 000~400 cm-1进行扫描[19]。
1.3.7 刚果红实验分析
分别称取5.0 mg三种多糖组分,加入2 mL蒸馏水,充分溶解后加入2 mL刚果红试剂(80 μmol/L),混匀;依次加入0.44、1.00、1.70、2.70、4.00 mL NaOH溶液(1 mol/L),NaOH最终为0、0.1、0.2、0.3、0.4和0.5 mol/L。于紫外-可见分光光度计中,在200~800 nm扫描,测量各NaOH浓度下溶液的最大吸收波长。不加多糖样品,按相同方法测量空白条件下的最大吸收波长[20]。
1.3.8 数据处理
实验数据均重复3次取平均值,使用Origin 8.5作图。
2 结果与分析
2.1 淡红侧耳粗多糖的分离纯化
2.1.1 DEAE-纤维素-52层析柱纯化
由图1可知,淡红侧耳粗多糖经DEAE-纤维素-52层析柱纯化后得到4个组分,分别为去离子水洗脱的组分PDP-1,0.1 mol/L NaCl洗脱的2个组分PDP-2和PDP-3以及0.3 mol/L NaCl洗脱的组分PDP-4。组分PDP-4得率过低,不易于收集,由于时间有限,故只收集前3个组分继续过Sephadex G-150层析柱。
图1 淡红侧耳粗多糖经DEAE-纤维素-52层析 柱纯化的洗脱曲线Fig.1 Elution curve of crude Pleurotus djamor polysaccharides on DEAE-cellulos-52 column
2.1.2 Sephadex G-150层析柱纯化
由图2可知,PDP-1、PDP-2和PDP-3经Sephadex G-150层析柱纯化后各得到一个组分,即PDP-1-1、PDP-2-1和PDP-3-1。这3个组分都呈现单一对称峰,表明所得组分为较纯多糖。分别收集这3个组分,50 ℃旋蒸浓缩,透析后真空冷冻干燥,得到白色絮状的淡红侧耳多糖纯品PDP-1-1、PDP-2-1和PDP-3-1。
2.2 淡红侧耳多糖的纯度及相对分子质量测定
由图3可知,淡红侧耳多糖纯化组分PDP-1-1、PDP-2-1、PDP-3-1的高效液相色谱图呈现单一对称峰,表明组分纯度较高。三组分的保留时间分别为17.175、17.309、13.956 min,根据标准曲线计算可得,三组分的相对分子质量分别为:12.9、10.6、2 753.7 kDa。
2.3 淡红侧耳多糖的单糖组成分析
由图4可知,单糖标品的保留时间如下:甘露糖(tR=16.030 min),核糖(tR=20.830 min),鼠李糖 (tR=21.700 min),葡萄糖醛酸(tR= 23.770 min),半乳糖醛酸(tR= 26.570 min),葡萄糖(tR=31.412 min),半乳糖(tR=35.426 min),木糖(tR=37.710 min),阿拉伯糖(tR=38.623 min),岩藻糖(tR=44.400 min)。纯化组分PDP-1-1由甘露糖(tR=16.011 min)、葡萄糖(tR=31.403 min)、半乳糖(tR=35.413 min)、阿拉伯糖(tR=38.944 min)、岩藻糖(tR=44.131 min)5种单糖组成,各单糖物质的量百分比分别为:4.79%、28.68%、47.59%、13.37%、5.57%;PDP-2-1由甘露糖(tR=16.155 min)、葡萄糖(tR= 31.424 min)、半乳糖(tR=35.179 min)、木糖(tR=37.611 min)、阿拉伯糖(tR=39.296 min)、岩藻糖(tR=43.819 min)6种单糖组成,各单糖物质的量百分比分别为:6.43%、33.65%、36.17%、7.20%、9.88%、6.68%;PDP-3-1由甘露糖(tR=16.366 min)、葡萄糖(tR= 31.307 min)、半乳糖(tR=35.019 min)、岩藻糖(tR=44.501 min)4种单糖组成,各单糖物质的量百分比分别为:4.12%、87.24%、3.90%、4.74%。

图2 PDP-1、PDP-2、PDP-3经Sephadex G-150层析柱纯化的洗脱曲线Fig.2 Elution curve of PDP-1、PDP-2、PDP-3 on Sephadex G-150 column

图3 淡红侧耳纯化多糖的高效液相色谱图Fig.3 HPLC of purified Pleurotus djamor polysaccharides
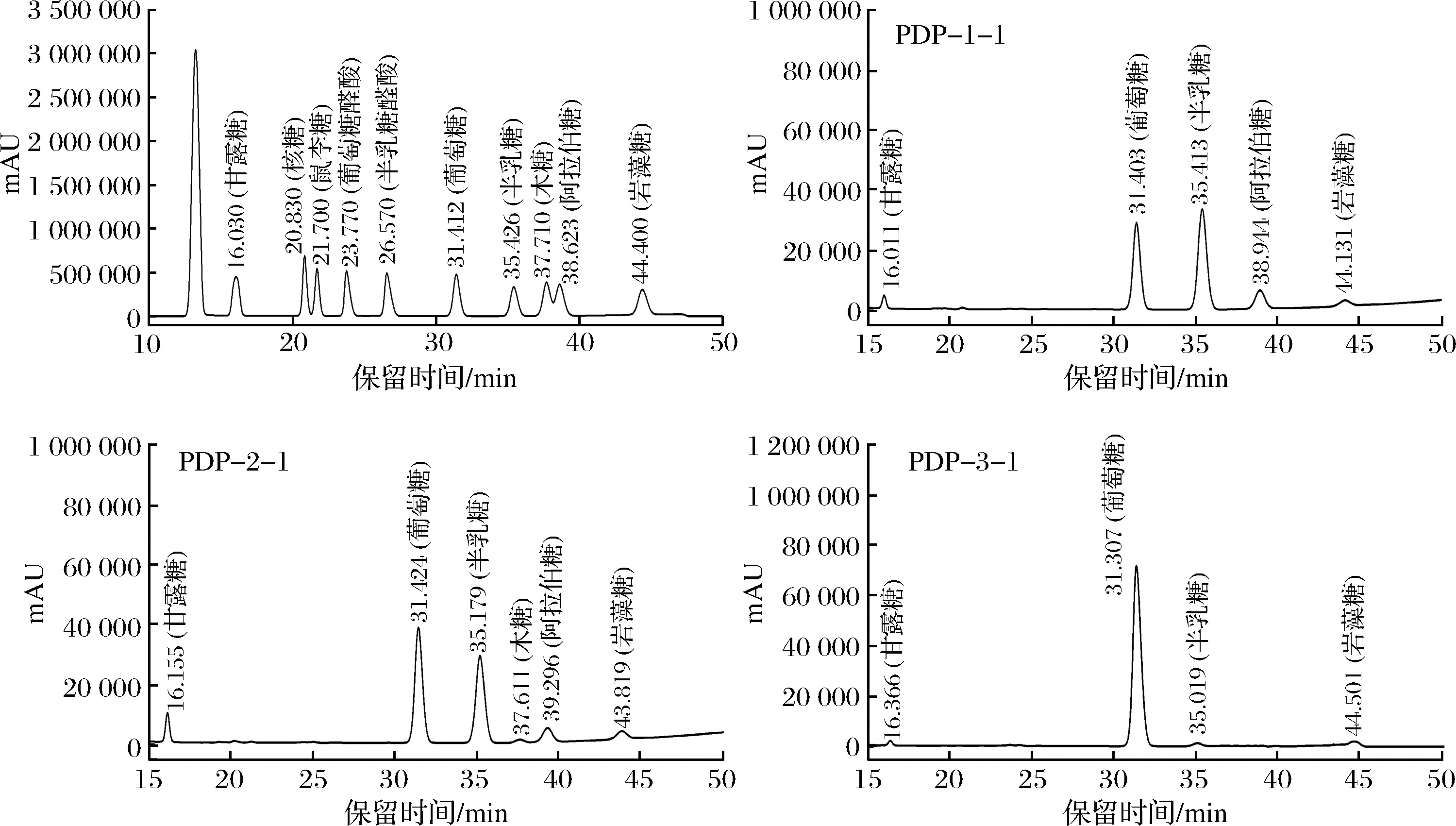
图4 混合单糖标准品及淡红侧耳纯化多糖的高效液相色谱图Fig.4 HPLC of mixed standard monosaccharide and purified Pleurotus djamor polysaccharides
2.4 理化性质的测定
总糖含量、蛋白质含量、糖醛酸含量、硫酸基含量的标准曲线分别为:y=0.007 4x+0.001 3(R2=0.999 5),y=0.003 5x-0.014 0(R2=0.998 4),y=0.012 5x+0.023 1(R2=0.999 3),y=0.001 7x-0.040 4(R2=0.999 6)。粗多糖及纯化组分中总糖、蛋白质、糖醛酸及硫酸基的具体含量见表1。由表1可知,与淡红侧耳粗多糖相比,纯化组分PDP-1-1、PDP-2-1和PDP-3-1的总糖含量明显增加;纯化组分的蛋白质含量减少,分别仅剩0.48%、0.79%、0.64%,这与紫外光谱分析结果一致;PDP-3-1的糖醛酸及硫酸基含量均高于粗多糖、PDP-1-1和PDP-2-1。有研究显示糖醛酸和硫酸基含量与多糖的生物活性呈正相关,乔德亮[21]从三角帆蚌中纯化出3种多糖组分HCPS-1、HCPS-2和HCPS-3,HCPS-3含有的糖醛酸和硫酸基均高于其他两组分,生物活性分析结果表明HCPS-3比HCPS-1和HCPS-2具有较高的体外抗氧化活性和免疫增强活性。
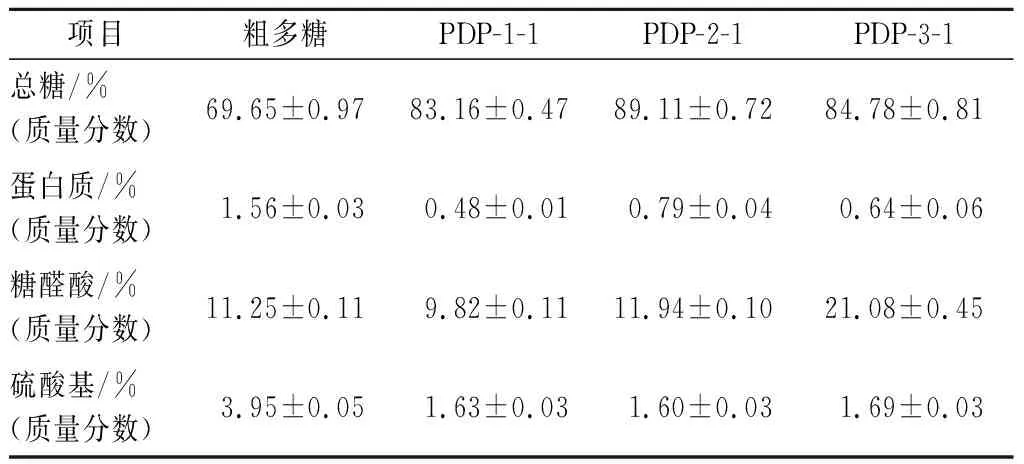
表1 淡红侧耳粗多糖及纯化多糖的理化性质Table 1 Physicochemical properties of crude and purified Pleurotus djamor polysaccharides
2.5 淡红侧耳多糖的光谱分析
2.5.1 紫外光谱分析
由图5可知,淡红侧耳粗多糖在260~280 nm有吸收峰,表明其含有蛋白质和核酸;纯化组分PDP-1-1、PDP-2-1和PDP-3-1在此处均无明显吸收峰,表明这3种纯化组分不含有或含有较少的蛋白质和核酸等物质,这与理化性质测定结果一致。

图5 淡红侧耳粗多糖及纯化组分的紫外光谱图Fig.5 Ultraviolet spectra of crude and purified Pleurotus djamor polysaccharides
2.5.2 红外光谱分析


图6 淡红侧耳多糖纯化组分的红外光谱图Fig.6 Infrared spectra of purified Pleurotus djamor polysaccharides
2.6 刚果红实验分析
刚果红是一种酸性染料,可与具有三股螺旋结构的物质发生络合作用,使溶液的最大吸收波长增加。当NaOH浓度较低时,溶液的最大吸收波长由于络合作用的发生而增大;NaOH浓度逐渐升高时,最大吸收波长由于三股螺旋的解开而减小。由图7可知,纯化组分PDP-1-1和PDP-3-1的最大吸收波长先增加后减小,说明其含有三股螺旋,而PDP-2-1的最大吸收波长一直呈减小趋势,说明其不含三股螺旋结构。

图7 NaOH浓度对淡红侧耳纯化多糖的影响Fig.7 Effect of NaOH concentration to purified Pleurotus djamor polysaccharides
3 结论
淡红侧耳粗多糖经DEAE-纤维素-52和Sephadex G-150纯化后共得到了3种组分PDP-1-1、PDP-2-1和PDP-3-1;采用高效凝胶过滤色谱法测定纯度及相对分子质量,可得三组分均为单一对称峰,说明纯化多糖的纯度较高;采用柱前衍生高效液相色谱法测定单糖组成,可知PDP-1-1主要由半乳糖、葡萄糖和阿拉伯糖等5种单糖组成,PDP-2-1主要由半乳糖和葡萄糖等6种单糖组成,PDP-3-1由葡萄糖和岩藻糖等4种单糖组成;紫外扫描和理化性质测定结果均表明3种纯化多糖含有极少量的蛋白质和较高的糖醛酸;红外扫描结果显示三组分均为吡喃糖,PDP-1-1和PDP-2-1以β-型糖苷键连接;刚果红结果显示PDP-1-1和PDP-3-1具有三股螺旋结构而PDP-2-1不具有三股螺旋结构。
多糖的结构与其生物活性密切相关,对多糖结构的深入研究,可推动多糖类新药的开发。目前对淡红侧耳多糖结构的解析只涉及特征官能团和单糖组成方面,为进一步了解糖苷键构型及单糖残基的连接方式等,还需进行甲基化和核磁共振分析,以期为其生物活性和构效间关系研究提供理论依据。
