电感耦合等离子体原子发射光谱(ICP-AES)法测定南红玛瑙中剧毒元素As、Cd、Cr、Pb、Sb的含量
2019-01-14邓传东安身平浦晨晨
邓传东 孙 琳 安身平 赵 峰 浦晨晨
(中国核动力研究设计院,成都 610213)
前言
近几年来,市场上出现了一些主要产于中国西南地区的南红玛瑙,因为天然形成的温润红色,正好迎合了我们中国人有关“中国红”的审美观,其也成为了许多玉石爱好者的新宠,成功吸引了大家的目光[1]。此外,南红玛瑙自古就有入药之风,有人认为其有特殊的药用功效。南红玛瑙主要矿物组成为石英,含有大量的Si,其次为Fe及少量的K、Ca、Al、Mg、Cl等,并且红色部分的鲜艳程度与Fe含量存在密切相关[2-3]。此外,其还含有V、Cr、Ni、Cu、Zn、Sr、V、Y、Zr、Mo、Ba、La、Ce、Nd、Pb等多种微量元素,且其元素含量存在较大差异。虽然各元素含量在不同样品之间存在较大差异,但总体都存在Cr、Pb富集现象[4]。
重金属元素As、Cd、Cr、Pb、Sb会对人类健康造成严重的影响,如我国化妆品相关标准规定了As、Cd、Cr、Pb、Sb等一些禁限用元素的含量与测定方法[5-6]。南红玛瑙作为一种中国人特别喜爱的饰品,且有时被人们磨成粉末制成浆液作药品吞服。因此,有必要建立南红玛瑙样品中重金属剧毒元素As、Cd、Cr、Pb、Sb的分析方法,为人们研究南红玛瑙各方面的价值提供参考。
电感耦合等离子体原子发射光谱(ICP-AES)法具备灵敏度高、精密度好、检出限低、线性范围宽、基体效应小、全谱直读等诸多优点,使用领域越来越广,在冶金、地质、环境、食品、中药、生化、石油化工等不同领域的应用越来越广泛[7-12]。目前还没有使用ICP-AES法测定南红玛瑙微量化学成分具体分析方法的报道,实验选择As、Cd、Cr、Pb、Sb具有典型的、具剧毒性的元素进行了分析研究[13-14]。使用硝酸与氢氟酸对南红玛瑙样品进行溶解,前处理简便,测定快速、准确,适用于南红玛瑙中As、Cd、Cr、Pb、Sb的测定。
1 实验部分
1.1 仪器与工作条件
iCAP 6300型全谱直读等离子体原子发射光谱仪(美国热电公司),配备耐氢氟酸型进样系统,等离子体频率27.12 MHz;射频功率1 150 W;辅助气流量0.50 L/min;雾化器气体流量0.55 L/min;泵速50 r/min;进样清洗时间25 s;观测方向为水平方向。
1.2 试剂与材料
聚四氟乙烯烧杯:50 mL;容量瓶:10、25、50 mL。
单元素标准溶液,具体浓度与标准编号见表1;混合标准溶液:使用单元素标准溶液配制混合标准溶液;硝酸、氢氟酸为优级纯,实验用水为超纯水(电阻率>18 MΩ·cm)。高纯氩气(Ar),纯度为99.999%以上。
南红玛瑙样品为有代表性的云南会泽南红玛瑙原石1个(见图 1a)。
所有玻璃器材与聚四氟乙烯器材均用HNO3溶液(10%)浸泡过夜,用水反复冲洗,最后用去离子水冲洗晾干使用。目标元素的分析线波长及使用的标准溶液相关信息见表1。

表1 元素分析线波长及标准溶液信息Table 1 Element analysis line wavelength and standard solution information
1.3 实验方法
1.3.1样品前处理
用去离子水与乙醇的混合溶液(V∶V=1∶1)洗涤南红玛瑙样品,晾干备用。敲碎已经洗涤好的南红玛瑙样品,称取1.0 g(精确至0.000 1 g)于聚四氟乙烯烧杯中,加入1 mL分析纯浓硝酸,逐步滴加氢氟酸直到样品完全溶解,冷却,用硝酸溶液(0.5%)定容至50 mL塑料容量瓶中,摇匀。
1.3.2测定方法
1.3.2.1标准曲线法
按照表2依次配制不同浓度的系列标准溶液,使用硝酸溶液(0.5%)进行定容。分别移取前处理过的样品试液5.0 mL于2个10 mL塑料容量瓶中,对于As、Cd、Cr、Sb元素,分别加入相应体积的标准溶液,定容,使这2份溶液的加入元素浓度分别为0.025、0.05 mg/L;对于Pb元素,分别移取处理好的样品试液1.0 mL于2个10 mL塑料容量瓶中,加入相应体积的标准溶液,定容,使其加入元素浓度分别为0.1、0.25 mg/L。测定样品中微量元素含量与加标回收量,并计算加标回收率。

表2 混合工作标准溶液系列浓度Table 2 Mass concentration of elements in working solution /(mg·L-1)
1.3.2.2 标准加入法
在实验中,分别移取处理过的样品试液5.0 mL于5个10 mL塑料容量瓶中,分别加入相应体积的混合标准溶液,定容。对于As、Cd、Cr、Sb元素,使这6份溶液(包括一份空白溶液)的加入元素浓度分别为0、0.025、0.05、0.1、0.25 mg/L。对于Pb元素,分别移取处理好的样品试液1.0 mL于5个10 mL塑料容量瓶中,加入相应体积的标准溶液,定容,使其加入元素浓度分别为0.1、0.25、0.50、1.0 mg/L。在ICP-AES仪上测量后由计算机绘制工作曲线,由工作曲线在X轴上的负截距即可得到试样中各元素的含量。
2 结果与讨论
2.1 样品的前处理选择
ICP-AES分析方法的准确性很大程度上取决于样品与标准溶液的匹配程度,所以样品称样量最好控制在合理范围内[11]。本实验称取1.0 g样品进行溶解以配制样品溶液。在样品溶解过程中,原本红色的样品在溶解完成之后得到的溶液呈无色透明状(如图1所示),可能是由于南红玛瑙中含有的Fe等致色元素含量较低的原因,溶解后颜色效应不明显。

a—破碎南红原石;b—碎样颗粒;c—接近溶解完成时的样品;d—溶解完成时的溶液
本实验使用氢氟酸与硝酸溶解南红玛瑙碎屑样品,在实验最后使用硝酸定容,以避免其中易水解元素沉淀析出。因此,考察了硝酸浓度变化对恒定目标元素含量测定的影响。固定基体溶液中As、Cd、Cr、Pb、Sb的浓度为0.2 μg/mL,在实验过程中配制不同硝酸浓度的溶液,做硝酸浓度对待测元素的干扰实验。从表 3可以看出,随着硝酸浓度增大,目标元素的检测值逐渐降低,其中Sb测定值的变化趋势相对比较平缓。这主要是由于硝酸浓度增大时,溶液的黏度随之增大而造成了吸液量减少,因此测定值逐渐降低。此外,当硝酸浓度大于1.0 mol/L时,检测值偏离程度较大,因此在实际实验中应该控制硝酸浓度小于1.0 mol/L。

表3 硝酸浓度变化对ICP-AES法测定0.2 μg/mL目标元素的影响Table 3 Influence of the concentration change of nitric acid on the determination of 0.2 μg/mL target elements by ICP-AES /(μg·mL-1)
样品的基体效应会对测定值产生一定的干扰,消除干扰的方法一般有扣除干扰系数法、基体匹配法及标准加入法,扣除干扰系数法受实验条件(介质、温度等)的影响较大,基体匹配法无法获得相应的基体来做匹配[15]。因此,本实验首先选择标准加入法来测定南红玛瑙中As、Cd、Cr、Pb、Sb微量元素,并比较了标准加入法与标准曲线法在分析测定结果方面的异同,进而得出适合于测定各微量元素的方法。
2.2 谱线选择与共存元素的干扰
南红玛瑙中的主体元素Si用氢氟酸溶解后生成气体SiF4可除去,对目标元素的测定影响不大。此外,南红玛瑙中还含有多种微量元素,且不同样品中其元素含量存在较大差异,含量较高的元素有Fe、Al、K、Ca、Mg等[2,4]。目标元素As、Cd、Cr、Pb、Sb都有多条发射谱线,由于谱线自身强度不同,不同元素之间存在一定的干扰。实验在未知样品元素含量情况下暂时选定高灵敏度的、无重叠的、稳定性好的、共存元素干扰小或者没有干扰的谱线作为测定谱线。通过被测元素光谱线的图形,观察干扰及背景影响情况,选择曲线光滑,信噪比高,附近干扰峰少的分析线[8]。经过选择,最终确定各元素的分析线,见表1。
2.3 校准曲线和方法检出限
在标准加入法中,加入恒定量的样品溶液,尔后加入相应体积的标准溶液定容摇匀后测定。在标准曲线法中,使用去离子水配制与表2相对应的浓度系列溶液并进行测定。标准加入法以目标元素标准溶液的加入浓度值为横坐标(x),标准曲线法以目标元素标准溶液的浓度值为横坐标(x),对应目标元素的分析线强度为纵坐标(y)绘制校准曲线。上述两种方法的线性范围、线性回归方程及相关系数见表 4。对试剂空白溶液连续测定10次,以3倍标准偏差(3s)作为方法的检出限,结果见表 4。由表 4可知,两种方法中各目标元素对应的相关系数均大于0.999,标准加入法的检出限在0.000 3~0.005 4 mg/L,标准曲线法的检出限在0.000 3~0.006 0 mg/L。对比相同元素的校准曲线之间的斜率可以发现,标准曲线法校准曲线的斜率总是大于标准加入法校准曲线斜率,由此可见基质对于目标元素校准曲线的斜率有明显的影响。

表4 线性参数与检出限Table 4 Linearity parameters and detection limits
2.4 方法精密度及加标回收实验
在样品中添加混合标准溶液,按实验方法进行加标回收实验,平行测定6次,结果见表 5。由表 5可知,在标准加入法中,各元素加标回收率在93.6%~103%,测定值的相对标准偏差(RSD)小于10%;在标准曲线法中,各元素加标回收率在90.2%~103%,各元素的相对标准偏差(RSD)也均小于10%,使用这两种方法测量各目标元素得到的相应RSD、加标回收率之间的差别并不明显。两种测定方法中As的RSD均比其它元素明显偏大。标准曲线法中Cd的回收率明显偏低,可能是因为基体效应造成发光强度降低而导致测定结果偏低。此外,标准加入法与标准曲线法的测定结果虽然相近,可也发现标准曲线法的测定结果总是比标准加入法的测定结果小,这再次说明由于标准曲线法中绘制标准曲线的溶液基体与实际测定样品的溶液基体存在差异,使得基体效应存在一定的影响,但不十分明显。标准加入法可以很好地减轻基体效应的干扰,但不可大批量测定样品。因此,在分析控制质量要求允许范围内,可采用标准曲线法对南红玛瑙中剧毒元素As、Cd、Cr、Pb、Sb的含量进行测定。
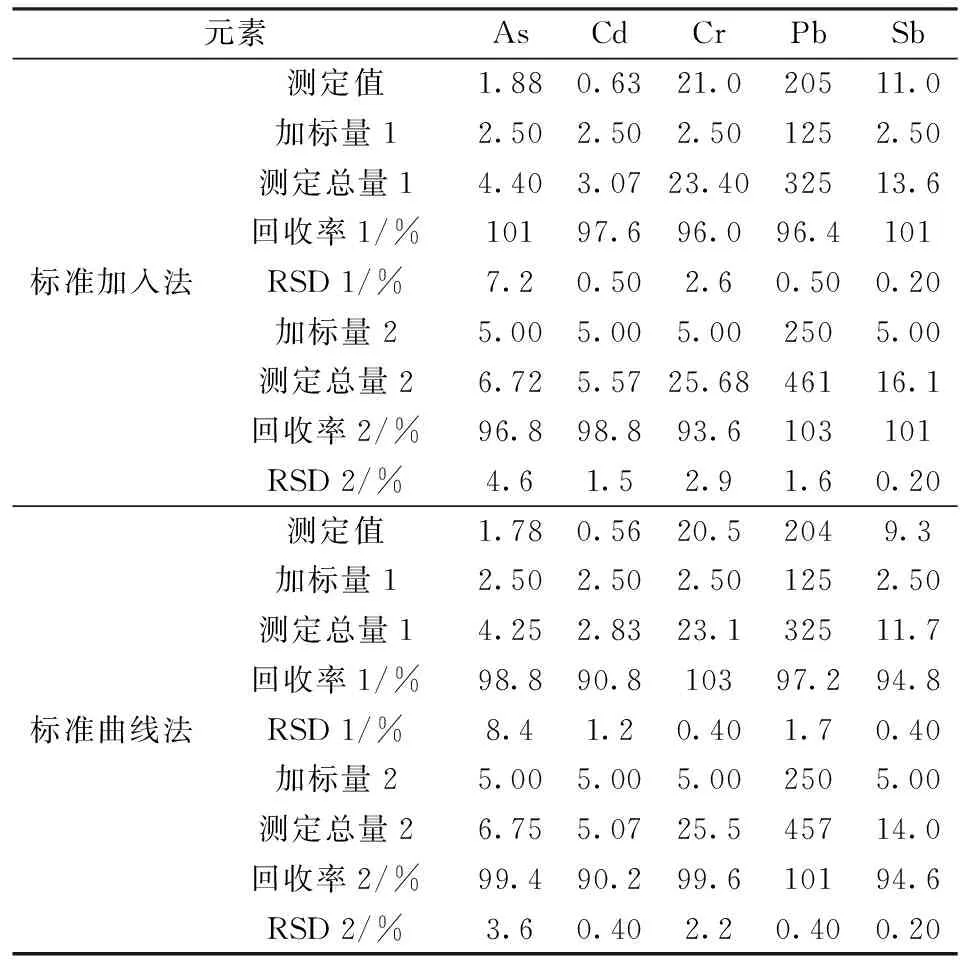
表5 方法精密度与加标回收实验Table 5 Results of tests for precision and recovery(n=6) /(μg·g-1)
从南红玛瑙中As、Cd、Cr、Pb、Sb测定的含量值来看,As、Cd的含量很低,Cr含量较高,Pb含量最高,且Pb含量高出了其它几种元素1~2个数量级。虽然本次取样的南红玛瑙产地与郭威等人实验分析的南红玛瑙产地不一致,但是分析结果能反应其提出的南红玛瑙总体上都存在Cr、Pb富集的现象[4]。此外,目前并没有确切的科学研究来证明南红玛瑙具有药用价值,但其中高含量的Pb所引发的生理健康问题值得引起人们的注意。
3 结论
采用硝酸-氢氟酸加热溶解南红玛瑙样品,并使用ICP-AES法测定南红玛瑙中的As、Cd、Cr、Pb、Sb含量。使用标准加入法和标准曲线法测量各目标元素得到的相应RSD、加标回收率之间的差别并不明显。在标准加入法中,各元素加标回收率在93.6%~103%,测定值的相对标准偏差(RSD,n=6)小于10%;在标准曲线法中,各元素加标回收率在90.2%~103%,各元素的相对标准偏差(RSD,n=6)也均小于10%。标准加入法中的回收率与精密度表明方法准确可靠,可同时满足南红玛瑙颗粒中As、Cd、Cr、Pb、Sb的分析要求。在分析控制质量要求允许范围内,可采用标准曲线法对南红玛瑙中剧毒元素As、Cd、Cr、Pb、Sb的含量进行测定。
