Photo-Induced Intermolecular Electron Transfer-Eflect of Acceptor Molecular Structures
2019-01-10WeiZhngXiosongLiuLinYnGngeiZhuZnhoWngYnqingYng
Wei Zhng,Xio-song Liu,Lin Yn,Gng-ei Zhu,Zn-ho Wng,Yn-qing Yng,∗
a.Department of Physics,Harbin institute of Technology,Harbin 150001,China
b.National Key Laboratory of Shock Wave and Detonation Physics,Institute of Fluid Physics,Chinese Academy of Engineering Physics,Mianyang 621900,China
Photo-induced electron transfer versus molecular structure of acceptors is investigated using ultrafast time-resolved transient grating spectroscopy.Typical laser dyes Rhodamine 101(Rh101)and Rhodamine 6G(Rh6G)in electron donor solvent—aniline are adopted as the objects.The forward electron transfer time constant from aniline to the excited singlet state of two Rhodamine dyes and subsequent back electron transfer from two dyes to aniline are measured.The experimental results denote that Rh6G presents faster electron transfer rates with aniline in both forward electron transfer and back electron transfer processes.With chemical calculation and qualitative analysis,it is found that the flexible molecular geometry of Rh6G leads to stronger electron coupling with donor solvent and further gives rise to larger electron transfer rates.
Key words:Electron transfer,Molecular structure,Back electron transfer
I.INTRODUCTION
Photo induced electron transfer(PIET)occurs in a wide range of fields,such as biological,photophysical and photochemical reactions[1,2],and the process is ultrafast and occurs in femtosecond or picosecond time scale[3].Relevant research is helpful to comprehend the essence of photosynthesis as well as to improve conversion efficiencies of solar cells.Owing to its scientific significance and tremendous application values,plenty of substantial investigations were conducted in both experimental and theoretical points of view[1−5].After Marcus developed the electron transfer(ET)theory,many factors which may aflect electron transfer(ET)rates are considered and discussed.The vibration of reactants[6,7],the solvent relaxation[8,9],the distance between donor and acceptor[10−12],the protocoupling[13,14]and other factors are examined.
Molecular structure is a fundamental aspect that determines the nature of matter. Diflerent structures would give rise to diflerent potential energy surfaces of reactants and diflerent inter-molecular electron coupling which are crucial factors in the ET reactions[15−17]. PIET has not been researched sufficiently in the past decades,Vauthey has researched the relationship between donor structure and the ET rates and found that with increasing substitution on the nitrogen atom the ET rate decreased[4].However,the structure eflect of acceptor on the ET rates has not been concerned.Revealing the structure eflects on the PIET process is helpful to promote comprehension of ET reactions and to improve photoelectric conversion efficiencies.
Rhodamine 101(Rh101)and Rhodamine 6G(Rh6G)in electron donating solvent aniline(AN)are used as the samples in this research.Geometries of the two dyes are similar,both include a xanthene ring and a phenyl ring,a number of nitrogen and oxygen atoms within the molecules are also equal.However,Rh6G owns more suspended groups like ethylamino groups and methyl substituents that link to the xanthene.So,unlike rigid Rh101,molecular structure of Rh6G is more flexible.Therefore,the two typical dye molecules provide suitable objects for studying the eflect of acceptor structure in PIET reactions.
PIET process can be illustrated in FIG.1. The laser dye and the electron donor(AN)molecule are both in electronic ground state at the beginning.After pump light stimulates the acceptor into electronic excited state,an electron jumps from the HOMO(highest occupied molecular orbital)to the LUMO(lowest unoccupied molecular orbital),a hole is generated in the HOMO.Then the excited dye would go through radiative transition and fluorescence is produced,the process lasts about 4 ns and the fluorescence quantum yields of two dyes are close to 1[18−20].While being dissolved in AN,an electron in the HOMO of AN would transfer into the hole in the HOMO of the stimulated dye and the fluorescence is quenched[21−23],which is FET pro-cess.Afterward,the electron in the LUMO of dye would go back to the HOMO of AN,which is the BET process.
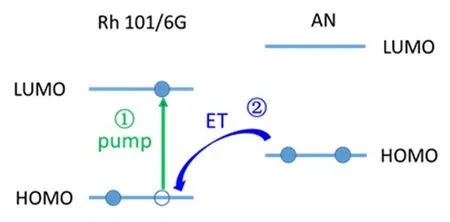
FIG.1 Schematic diagram of PIET process.A pump pulse stimulates the Rhodamine molecule into the excited state.A hole emerges in the HOMO.Then,an electron from HOMO of AN transfers to the hole and fluorescence of excited dye is quenched.
Transient grating(TG)spectroscopy[5,24,25]is a four wave mixing technique.As illustrated schematically in FIG.2,two spatially crossed and timecoincident pump pulses,with the same central wavelength but diflerent wave vector directions are employed.Interference pattern would be generated in the overlapping region in the sample.While a third,timedelayed probe light shots on the grating,TG signal that contains dynamical information of the sample would appear in the phase-matching direction.As the TG signal is background free,so its signal-to-noise ratio is extremely high.Previous researches have been proven that TG spectroscopy is an appropriate technique to research the inter-molecular ET between Rhodamines and electron donor solvents[21,25,26].TG signal’s square root is proportional to the specific populations[4,27].
In this work,two typical electron acceptors(Rh101 and Rh6G)are adopted,each molecule has its own structural characteristics.So,by using the same electron donor,through contrastive experiments,the eflect of flexible molecular structure of electron acceptor on the PIET reactions can be revealed by time-resolved TG spectroscopy.
II.SAMPLE PREPARATION AND EXPERIMENTS
Rh101 and Rh6G are typical xanthene derivatives,they are usually used as active medium in dye lasers and as standard samples due to their high fluorescence quantum yields. Rh101 and Rh6G were purchased from Sigma-Aldrich.They were dissolved in AN with the same concentrations of 0.2 mmol/L,and the two samples would exist in the forms of cation(Rh101+,Rh6G+)in solvents.The sample solutions were placed in a 2-mm quartz cell in the experiments.
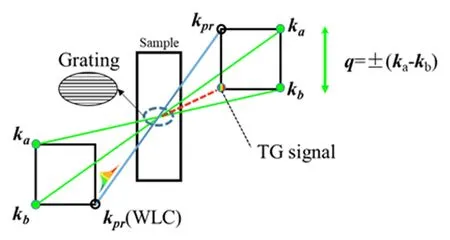
FIG.2 Beam geometry for broadband TG experiment.
Steady-state absorption spectra were measured with an UV-VIS spectrophotometer(720PC,Shanghai).Steady-state fluorescence spectra of two molecules were measured in the nonreactive solvent methanol(MeOH)with a spectrometer(Chromex 500IS/SM,BRUKER),the central wavelength of excitation laser was 532 nm.AN and MeOH were of analytical reagent grade and used without further purification.Neither MeOH nor AN is electronic resonant with the excitation pulse in TG measurement.
Broadband TG technique has been reported previously[4,23,28].In brief,the 800 nm,110 fs pulse duration,1 kHz repetition output pulse from spitfire(Spectra Physics)was used as the primary beam,it was split into two beams by a 9:1 beam splitter.The 90%beam pumped an optical parametric amplifier(OPA,OPA-800FC,Spectra Physics)to produce a pulse with a central wavelength of 532 nm and a full-width at 1/e maximum about 10 nm,which was branched by a 1:1 beam splitter to produce two equal pump pulses in the experiments.The other 10%beam was focused onto water to produce a white light continuum(WLC)which was utilized as the probe in TG measurement.The Kerr eflect was measured to calibrate the eflect of the group velocity dispersion of the WLC[25].All three beams were aligned parallelly to one another and spatially overlapped at the common focus in the sample by the achromatic lens with the focal length of 300 mm.The intersection angle of two pump pulses was about 2.86◦.
Beam geometry and photograph of TG signal is shown in FIG.2.kaand kbare pump pulses,kcrepresents probe beam.Forward box arrangement was chosen for better signal-to-noise ratio[29].The background free signal was gathered in ks=ka−kb+kcdirection.Relative timing between the two pump pulses were fixed to be overlapped while the probe pulse was time-scanning by means of a computer-controlled stepping motor with a minimal step size of 20.83 fs.The TG signal was recorded by a spectrometer(Avantes,PMC-100)in phase-matching direction.
In experiments,polarization direction of the pump and probe pulses were scheduled horizontally.Zero time point is defined as the point when the pump and probe pump crossed in the sample.Positive delay time represents pump pulses preceding the probe,and inverse negative.All experiments were carried out at room temperature(23◦C)in dark room.

FIG.3(a)Normalized steady-state absorption and fluorescence spectra of Rh101+/AN and Rh101+/MeOH.(b)Normalized steady-state absorption and fluorescence spectra of Rh6G+/AN and Rh6G+/MeOH.Insets are the molecular skeletons of Rh101 and Rh6G respectively.
III.RESULTS AND DISCUSSION
A.Steady-state UV-Vis absorption and fluorescence spectra
Steady-state UV-Vis absorption spectra of Rh101+and Rh6G+in AN and fluorescence of Rh101+and Rh6G+in MeOH were measured,the normalized spectra are shown in the FIG.3.As the fluorescence of two dyes was quenched by AN,the fluorescence spectra of two dyes were measured in the non-reactive solvent MeOH.
Normalized steady-state absorption of Rh101+/AN and Rh6G+/AN are shown as black lines that are marked with squares in FIG.3,the peaks locate around 574 nm and 540 nm respectively.Both of the two curves show vibronic shoulders at the blue side of the peak,which are derived from in-plane deformation of the xanthene ring[30,31].The fluorescence of Rh101+and Rh6G+in MeOH are quite strong,normalized spectra are featured by peaks at 593 nm and 555 nm,respectively,shown as red curves that are marked with red triangles.
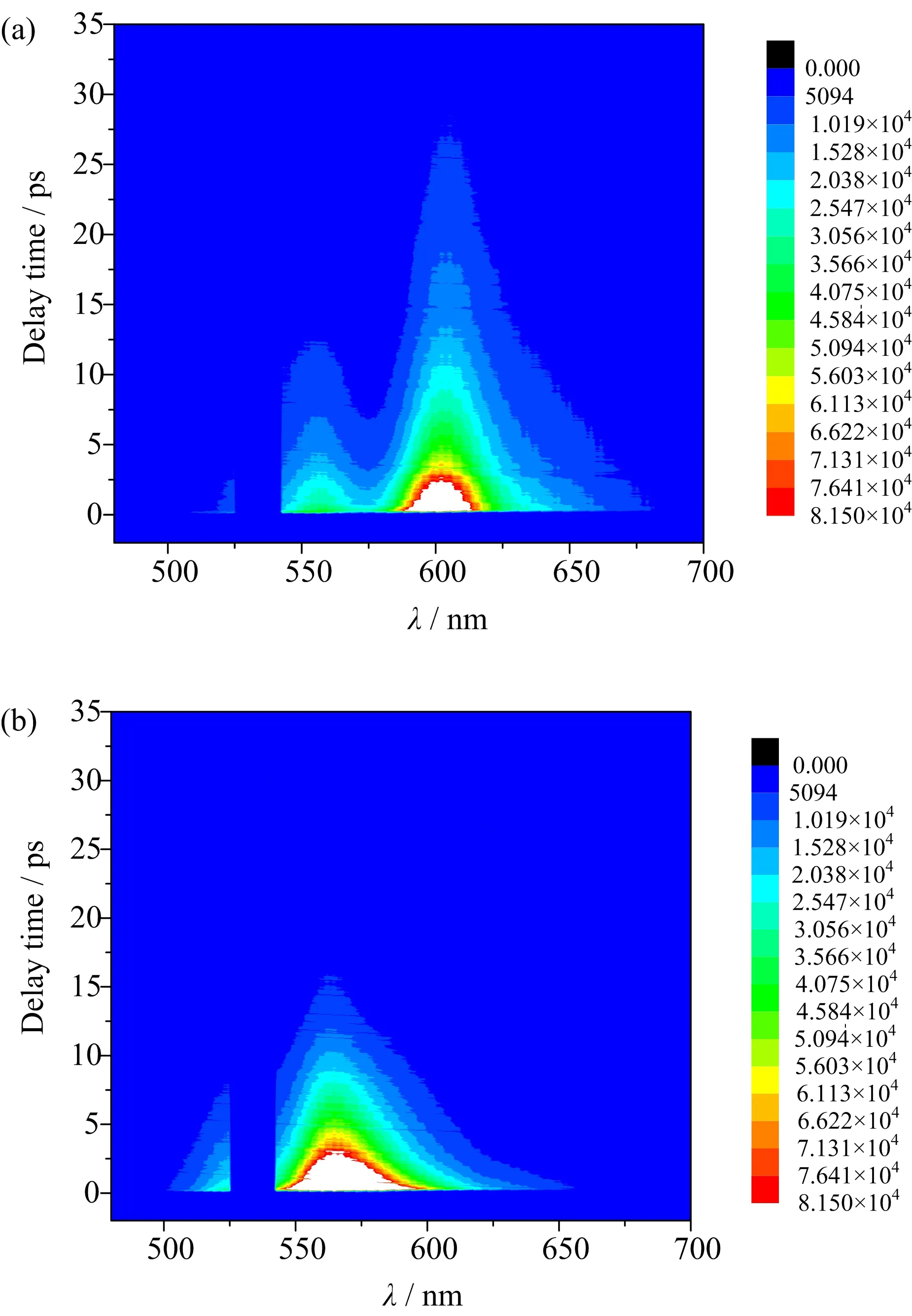
FIG.4 Time and frequency resolved TG spectra of(a)Rh101/AN and(b)Rh6G/AN.
B.Experimental measurements of PIET by TG spectroscopy
Time-resolved broad band TG experiments were carried out to track the ultrafast FET processes and subsequent BET processes.The measured TG spectra of the two samples are shown in FIG.4,the horizontal axis presents wavelength and the vertical axis denotes relative delay time between probe pulse and pump pulses.To eliminate the intense scattered pump light,a notchfilter with central wavelength at 532 nm was used in the signal collection light path so the signal between 525−542 nm band was blank in the figure.
As the fluorescence is quenched by intermolecular electron transfer from AN to Rh101+∗,the stimulated emission(SE)quenching rate corresponds to ET rate.In the absorption band of the sample,ground state bleach(GSB)evolution is contained,it reflects BET dynamics.The concepts of SE and GSB are borrowed from transient absorption.In the TA measurement,a pump light stimulates the sample into electronic excited state.Afterward,a broad band probe light strikes on the sample,the SE and GSB would occur.In the TG measurement,the experienced process of the sample is the same as that in TA,and the evolutionary information of the sample is uniform at the same detecting wavelength in both TA and TG measurements.
According to the steady-state fluorescence spectra of the two samples,the peaks of SE of Rh101+/MeOH and Rh6G+/MeOH are 593 and 555 nm.However,at the two wavelengths,the absorbance of the two samples are not zero.To avoid the eflect of ground state bleach,the detecting wavelengths are chosen at pure SE region.Specifically,FET dynamics of Rh101+/AN is measured at 630 nm while FET dynamics of Rh6G+/AN is measured at 610 nm,the selected wavelengths are labelled as red dashed arrows in the fluorescence spectra in FIG.3.
To ensure the reliability of the comparison,the FET dynamics is evaluated versus laser power in two cases.Comparison of FET dynamics with diflerent pump photon flux of the two samples is shown in FIG.5(a)and(b).Laser powers of pump pulse are chosen to be 0.05,0.10,and 0.15 mW respectively.Vertical axis shows the square root of TG intensities in semi-log scale,and horizontal axis expresses the delay time.
To achieve a more reasonable comparison,the TG dynamics is normalized,as shown clearly in FIG.5.No matter Rh101+/AN or Rh6G+/AN,the ET dynamical curves are almost overlapped at diflerent light energy of pump light.It indicates that,in the energy range that we choose,ET rates are independent of the intensity of the excitation light.Theoretically,the larger the pump laser intensity is,the more population of Rhodamine dye would be stimulated into electronic excited state.In statistical terms,the quantity of population does not aflect the evolution dynamics.Experimental results denote that there is no more unknown eflect that is caused by the laser intensity which can aflect PIET reactions.
The PIET processes are not strictly exponential and are consistent with previous reports[22,26].In order to interpret the diflerences detected in two cases,it’s necessary to fit the experimental data to extract more information.The intermolecular ET can be regarded as two compositions roughly,a faster one at the beginning and a slower one subsequently.The fast ET components are originated from donor and acceptors with optimal orientations,the slower ones are those with non-optimal orientations[32].
A bi-exponential equation is employed to fit the experimental data at diflerent wavelengths during fluorescence decay periods.The fitting equation is written as follows:

here t1,t2are two lifetime components,and A1,A2represent the corresponding weight constant.The bestfi t parameters corresponding to two samples are listed in Table I.
It has also been reported that ET time from N,NDMA and N,N-DEA to the excited state of dyes oxazine-1,Rh101,Rh6G varies from dozens to hundreds of femtosecond[22,32,33],such a rapid ET process can be regarded as barrierless[26].However,the ET process from AN to Rh101+∗and Rh6G+∗lasts about morethan 10 ps.The relative slow ET rate suggests that the electron donating ability of AN is relative weak and there is a potential barrier between ET reactants and products in this work.
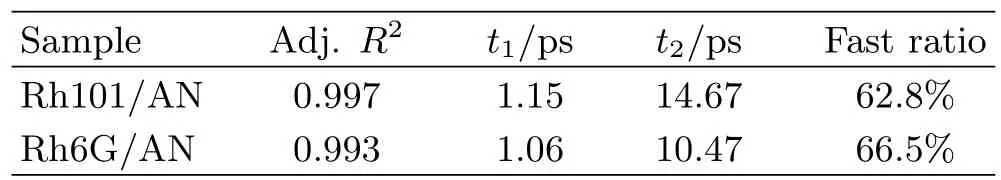
TABLE I Best-fit PIET time constant and fast components proportion measured by TG.
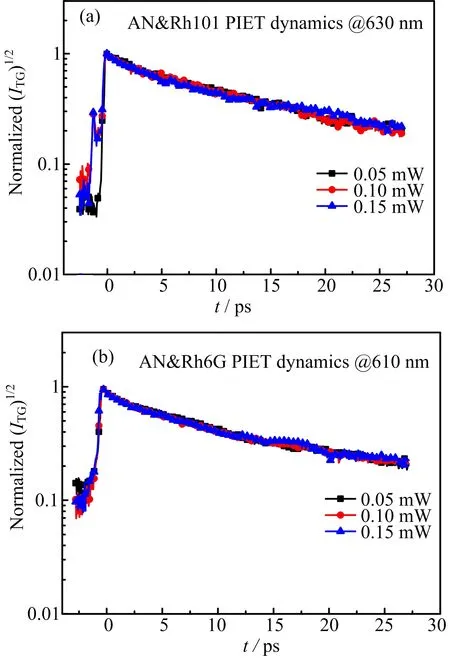
FIG.5 Normalized PIET dynamicscomparison of(a)Rh101/AN and(b)Rh6G/AN versus diflerent pump powers.The PIET dynamics of the two samples shows almost invisible dependence on the power of pump laser.
The Adj.R2shows the coefficient of determination between original data and the fitted data.The constants in two cases are both greater than 0.99 which denotes the fitting degree is reasonable.With the system of Rh101/AN,there is a fast time constant about(1.15±0.09)ps and a slow one about(14.67±0.31)ps.While in the Rh6G/AN system,the fast and slow time constants are(1.06±0.07)ps and(10.47±0.15)ps,respectively.In both cases,the fast time constant is predominant,which occupies more than 60%.ET rates vary inversely with the time constants.So,obviously,ET rate between Rh6G+and AN is faster than that between Rh101+and AN.

FIG.6 BET dynamics of(a)Rh101/AN and(b)Rh6G/AN at diflerent photo fluxes of pump light.
The BET of two samples are also evaluated by the TG spectroscopy,as shown in FIG.6.After FET,the reaction intermediate Rh101·∗and Rh6G·∗(neutral radical)and AN+would go through BET(also known as charge recombination)[4,22]subsequently.Thus population of the ground state Rh101+would increase and the diflracted TG signal in the absorption band would decrease.Therefore,the TG dynamics in absorption band would include BET dynamics.The detecting wavelengths of BET of the two sample are selected at 540 nm and 510 nm respectively which are in pure absorption band,marked as black arrows in the steady-state absorption spectra in FIG.3.
Spikes,which are caused by the optical Kerr eflect,emerge,but the eflect is only aflected at first few hundreds of femtosecond.So the normalization of the dynamical curves avoid the spikes.On the whole,the TG dynamics of each sample is almost overlapped with different photo flux of pump pulse.That denotes BET process is also independent of the laser power.BET presents single-exponential decay tendency,a single exponential equation is adopted to fit the curves.The coefficients of determination in two cases are very close to 1.As summarized in Table II,the fitting results indicates that BET between Rh101·∗and AN+lasts about(10.01±0.08)ps while the BET time constant between Rh6G·∗and AN+is about(8.11±0.04)ps.BET rate between Rh6G·∗and AN+is still larger than that of between Rh101·∗and AN+.
The FET and BET ratio diflerence between Rh101/AN and Rh6G/AN system can be well inter-preted by classic Marcus theory which is given by:
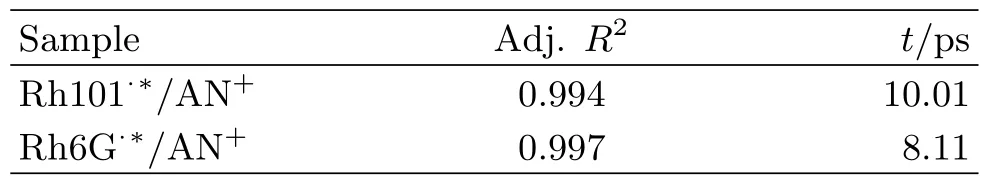
TABLE II Best-fit BET time constant measured by TG.
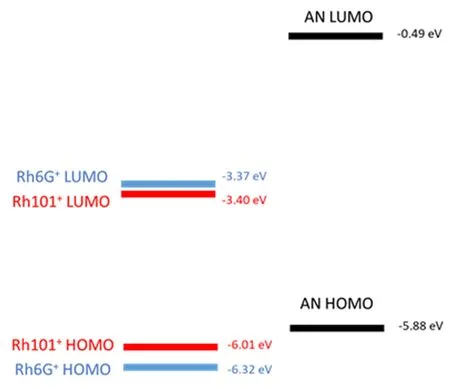
FIG.7Calculated HOMO and LUMO energy levels of Rh101+,Rh6G+,and AN.

HDAis the electron coupling strength between the donors and acceptors.λ denotes the whole reorganization energy which includes solvent reorganization energy and intramolecular reorganization energy.∆G0is the driving force of ET that represents the free energy diflerence between donor and acceptor.In the normal region,larger driving force makes larger ET rates.On the contrary,in the inverted region,larger driving force leads to smaller ET rate.It has been proven that the FET reaction between aniline and dyes is in the Marcus normal region while the BET occurs in the inverted region[22,34].
As Rh101 and Rh6G have their own molecular characteristics,the key factors in the ET reactions are quite diflerent.To begin with,the driving force is diflerent.The HOMO and LUMO energy of AN and the two dyes are calculated by Gaussian 03[35]program with B3LYP method and 6-311+G(d,p)basis set.Aniline is selected as the solvent in all the three cases.The calculated energy levels of the reactants are illustrated in FIG.7.The ionization energy of electron donor is identical in two cases,however,the HOMO energy of Rh6G+is 0.29 eV lower than that of Rh101+,so the driving force between Rh6G+and AN is larger than that between Rh101+and AN.That’s a main reason why ET occurs faster between Rh6G+and AN.
In addition,the molecular structure of Rh6G diflers from Rh101 in several aspects.As illustrated in the insets of FIG.3,Rh6G holds more pendent groups.For instance,there are two symmetrical ethylamino groups and methyl substituents on the xanthene ring. Besides,the carboxyl group on the phenyl ring is esterified.Among these key factors in the Marcus ET equation,HDAis proportional to the geometry of the reactants.In a quantum mechanics perspective,HDAcan be defi ned as related to the forward electron orbital overlap of the donor and the acceptor.
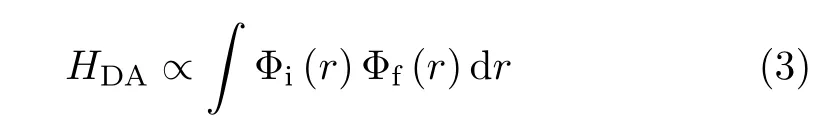
Φirepresents the initial orbital wave function of the electron and Φfis the final orbital wave function in the ET process.In the FET process,electron transfers from AN’s HOMO to the Rhodamine dye’s HOMO.On the contrary,in the BET process,electron transfers from LUMO of Rhodamine dye to the HOMO of AN.
Compared with Rh101+,Rh6G+holds higher LUMO level.In view of BET occurring in the inverted region,the larger driving force may cause smaller BET rate.However,the measured BET rate of Rh6G+/AN is about 20%larger than that of Rh101+/AN.The driving force diflerence between two dyes and AN is only close to 1%,the tiny diflerence has little influence on BET rates.Therefore,it can be concluded that the electron coupling between donor and acceptor plays a primary role in BET reactions.
In view of the formula of HDA,the greater overlap of initial orbit in electron donor and final orbit in electron acceptor is,the larger ET coupling coefficient[36−38].As the electron donor is selected as AN in two cases,compared with Rh101,Rh6G holds more pendent groups.The wave function overlap between Rh6G and AN is larger than that between Rh101 and AN,which means flexible molecular structure of Rh6G has greater coupling coefficients with adjacent ANs.That is also a reason that Rh6G holds larger FET rate with AN than Rh101.
IV.CONCLUSION
Ultrafast PIET reactions of two typical Rhodamine dyes in pure donor solvent(AN)are investigated by TG spectroscopy.The ultrafast FET and subsequent BET reactions between two dyes and AN are evaluated versus pump laser power.The results show that the normalized FET and BET dynamics are almost overlapped with diflerent photo flux,which denotes the ET rates are immune to the laser power within the selected power range.FET and BET time constants are also obtained through the TG dynamics at diflerent wavelengths.It is found that the Rh6G presents faster ET rates with AN in both FET and BET processes.Through quantum chemical calculation and qualitative analysis,it can be inferred that molecular structure diflerence leads to different driving force and electron coupling between the reactants.Specifically,flexible molecular structure of acceptors gives rise to greater electron coupling with donors and further larger ET rates in both FET and BET reactions.
The measured FET and BET time constants provide referential significance in the relevant research.Furthermore,the revealed ET rates diflerence caused by specific molecular skeleton is helpful in improving photoelectric conversion efficiency and in designing and manufacturing photovoltaic devices.
V.ACKNOWLEDGMENTS
This work was supported by the Science Challenge Project(No.TZ2016001),and the National Natural Science Foundation of China(No.21673211).
[1]R.A.Marcus and N.Sutin,BBA-Rev Bioenergetics 811,265(1985).
[2]M.D.Newton and N.Sutin,Annu.Rev.Phys.Chem.35,437(1984).
[3]A.Rosspeintner,B.Lang,and E.Vauthey,Annu.Rev.Phys.Chem.64,247(2013).
[4]E.Vauthey,J.Phys.Chem.A 105,340(2001).
[5]T.Kumpulainen,B.Lang,A.Rosspeintner,and E.Vauthey,Chem.Rev.117,10826(2017).
[6]Y.Shim,D.Jeong,S.Manjari,M.Y.Choi,and H.J.Kim,Acc.Chem.Res.40,1130(2007).
[7]J.Sukegawa,C.Schubert,X.Z.Zhu,H.Tsuji,D.M.Guldi,and E.Nakamura,Nat.Chem.6,899(2014).
[8]I.Rips and J.Jortner,Chem.Phys.Lett.133,411(1987).
[9]M.Bixon and J.Jortner,Chem.Phys.176,467(1993).
[10]N.S.Hush,Coord.Chem.Rev.64,135(1985).
[11]G.L.Closs and J.R.Miller,Science 240,440(1988).
[12]G.B.Schuster,Acc.Chem.Res.33,253(2000).
[13]A.E.F.Nassar,Z.Zhang,N.F.Hu,J.F.Rusling,and T.F.Kumosinski,J.Phys.Chem.B 101,2224(1997).
[14]R.I.Cukier and D.G.Nocera,Annu.Rev.Phys.Chem.5,7695(2012).
[15]L.Chen,R.Durley,B.J.Poliks,K.Hamada,Z.Chen,F.S.Mathews,V.L.Davidson,Y.Satow,E.Huizinga,and F.M.Vellieux,Biochem.31,4959(1992).
[16]G.Kurisu,M.Kusunoki,E.Katoh,T.Yamazaki,K.Teshima,Y.Onda,Y.Kimataariga,and T.Hase,Nat.Struct.Mol.Biol.8,117(2001).
[17]M.G.Bertero,R.A.Rothery,M.Palak,C.Hou,D.Lim,F.Blasco,J.H.Weiner,and N.C.Strynadka,Nat.Struct.Mol.Biol.10,681(2003).
[18]T.Karstens and K.Kobs,J.Phys.Chem.84,1871(1980).
[19]R.F.Kubin and A.N.Fletcher,J.Lumin.27,455(1982).
[20]D.Magde,R.Wong,and P.G.Seybold,Photochem.Photobiol.75,327(2010).
[21]L.L.Jiang,W.L.Liu,Y.F.Song,X.He,Y.Wang,C.Wang,H.L.Wu,F.Yang,and Y.Q.Yang,Chem.Phys.429,12(2014).
[22]V.O.Saik,A.A.Goun,and M.D.Fayer,J.Chem.Phys.120,9601(2004).
[23]A.Morandeira,A.Furstenberg,and E.Vauthey,J.Phys.Chem.A 108,8190(2004).
[24]H.Kandori,K.Kemnitz,and K.Yoshihara,J.Phys.Chem.96,8042(1992).
[25]L.L.Jiang,W.L.Liu,Y.F.Song,X.He,Y.Wang,and Y.Q.Yang,Appl.Phys.B 116,271(2014).
[26]Q.H.Xu,G.D.Scholes,M.Yang,and G.R.Fleming,J.Phys.Chem.A 103,10348(1999).
[27]J.Etchepare,G.Grillon,J.P.Chambaret,G.Hamoniaux,and A.Orszag,Opt.Commun.63,329(1987).
[28]G.Y.Yu,Y.F.Song,Y.Wang,X.He,Y.Q.Liu,W.L.Liu,and Y.Q.Yang,Chem.Phys.Lett.517,242(2011).
[29]J.A.Shirley,R.J.Hall,and A.C.Eckbreth,Opt.Lett.5,380(1980).
[30]S.Shim,C.M.Stuart,and R.A.Mathies,ChemPhysChem 9,697(2008).
[31]H.Watanabe,N.Hayazawa,Y.Inouye,and S.Kawata,J.Phys.Chem.B 109,5012(2005).
[32]I.V.Rubtsov,H.Shirota,and K.Yoshihara,J.Phys.Chem.A 103,1801(1999).
[33]S.Engleitner,M.Seel,and W.Zinth,J.Phys.Chem.A 103,3013(1999).
[34]H.L.Tavernier,M.M.Kalashnikov,and M.D.Fayer,J.Chem.Phys.113,10191(2000).
[35]M.J.Frisch,G.W.Trucks,H.B.Schlegel,G.E.Scuseria,M.A.Robb,J.R.Cheeseman,J.A.Montgomery Jr.,T.Vreven,K.N.Kudin,J.C.Burant,J.M.Millam,S.S.Iyengar,J.Tomasi,V.Barone,B.Mennucci,M.Cossi,G.Scalmani,N.Rega,G.A.Petersson,H.Nakatsuji,M.Hada,M.Ehara,K.Toyota,R.Fukuda,J.Hasegawa,M.Ishida,T.Nakajima,Y.Honda,O.Kitao,H.Nakai,M.Klene,X.Li,J.E.Knox,H.P.Hratchian,J.B.Cross,C.Adamo,J.Jaramillo,R.Gomperts,R.E.Stratmann,O.Yazyev,A.J.Austin,R.Cammi,C.Pomelli,J.W.Ochterski,P.Y.Ayala,K.Morokuma,G.A.Voth,P.Salvador,J.J.Dannenberg,V.G.Zakrzewski,S.Dapprich,A.D.Daniels,M.C.Strain,¨O.Farkas,D.K.Malick,A.D.Rabuck,K.Raghavachari,J.B.Foresman,J.V.Ortiz,Q.Cui,A.G.Baboul,S.Cliflord,J.Cioslowski,B.B.Stefanov,G.Liu,A.Liashenko,P.Piskorz,I.Komaromi,R.L.Martin,D.J.Fox,T.Keith,M.A.Al-Laham,C.Y.Peng,A.Nanayakkara,M.Challacombe,P.M.W.Gill,B.Johnson,W.Chen,M.W.Wong,C.Gonzalez,and J.A.Pople,Gaussian 03(Revision B.01),Pittsburgh,PA:Gaussian,Inc.,(2003).
[36]M.J.Ondrechen and M.A.Ratner,Chem.Phys.Lett.51,573(1977).
[37]P.M.M.Gabas,L.F.Errea,L.Mendez,and I.Rabadan,Phys.Rev.A 85,799(2012).
[38]I.V.Kurnikov and O.V.Gritsenko,Chem.Phys.Lett.185,365(1991).
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Imaging HNCO Photodissociation at 201 nm:State-to-State Correlations between CO(X1Σ+)and NH(a1∆)
- Energy-Transfer Processes of Xe(6p[1/2]0,6p[3/2]2,and 6p[5/2]2)Atoms under the Condition of Ultrahigh Pumped Power
- Ultrafast Investigation of Excited-State Dynamics in Trans-4-methoxyazobenzene Studied by Femtosecond Transient Absorption Spectroscopy
- Strong Current-Polarization and Negative Diflerential Resistance in FeN3-Embedded Armchair Graphene Nanoribbons
- Unexpected Chemistry from the Homogeneous Thermal Decomposition of Acetylene:An ab initio Study
- Direct Observation of Transition Metal Dichalcogenides in Liquid with Scanning Tunneling Microscopy