Unexpected Chemistry from the Homogeneous Thermal Decomposition of Acetylene:An ab initio Study
2019-01-10EndongWngGungyueLiJunxiDingGuozhongHe
En-dong Wng,Gung-yue Li,Jun-xi Ding,Guo-zhong He
a.State Key Laboratory of Molecular Reaction Dynamics,Dalian Institute of Chemical Physics,Chinese Academy of Science,Dalian 116023,China
b.University of the Chinese Academy of Sciences,Beijing 100049,China
c.College of Chemical Engineering,North China University of Science and Technology,Tangshan 063009,China
The formation of the aromatic ring during the formation of polycyclic aromatic hydrocarbons(PAHs)remains controversial and the experimental evidence is still lacking.Moreover,the formation mechanism of benzene from acetylene in the gas phase has also puzzled organic chemists for decades.Here,ab initio molecular dynamics simulations and electronic structure calculations provide compelling evidence for an unexpected competitive reaction pathway in which the aromatic ring is formed through successive additions of vinylidene.Moreover,no collisions cause bond dissociation of the acetylene molecule during the formation of benzene in this work.This study reveals the key role for the vinylidene carbene and determines the lifetime of vinylidene.
Key words:ab initio calculations,Acetylene,Combustion,Aromatic ring,Carbenes
I.INTRODUCTION
Numerous reactions proceed rapidly and exothermically in combustion processes.Among them,reactions leading to PAHs have attracted considerable attentions because PAHs,which are by-products of the burning of fossil fuels,cause many environmental problems[1,2].Furthermore,reactions leading to aromatic compounds in the gas phase have long been of interest to organic chemists since Berthelot succeeded to synthesize benzene from acetylene in 1867[3].Thus,a clear understanding of the formation of PAHs is crucial to reduce the number of PAH-like compounds in the combustion emissions and will also benefit organic chemists.
The hydrogen abstraction/acetylene addition[4–7]mechanism has been considered a key route for the formation of PAHs by a number of researchers[7–12].Notably,a common basis of these research indicates that an aromatic ring must be present to initialize the subsequent HACA reactions.However,diflerences emerge regarding how the ring is formed.In essence,the disagreements involve three types of reactions[3,13–15].One type of formation reaction is the even-carbon-atom pathway,which involves addition reactions between C4 hydrocarbons and C2 hydrocarbons[5,16–18]including
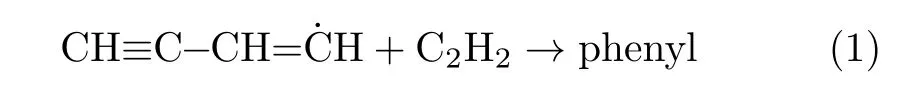
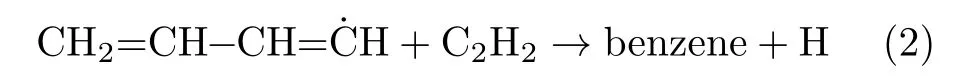
Another type of formation reaction is the odd-carbonatom pathway[19–21]:

Finally,the third type,which is discussed below,is the cyclization pathway[14]:
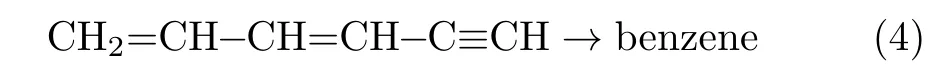
Although several studies support reactions(1)−(3),evidence to dismiss these pathways also exists.For instance,there are uncertainties regarding the concentrations of n-C4H3and n-C4H5for the even-carbonatom pathway[22].Furthermore,for the odd-carbonatom pathway,the experimental reactants include 1,5-hexadiyne[21],allene[19],and Na+C3H3X[20](X=Cl or Br).Thus,further investigation is required to clarify the formation of the aromatic ring.Moreover,although significant progress has been made in the development of scientific instruments,it remains difficult to monitor complex chemical reactions,such as the ultrafast reactions that occur during combustion process,and to detect short-lived intermediates in situ.To the best of our knowledge,this reason is primarily why the formation route for the aromatic ring in the HACA mechanism has not yet been directly observed experimentally.Ab initio molecular dynamics simulations are considered suitable to compensate for the lack of experimental data.
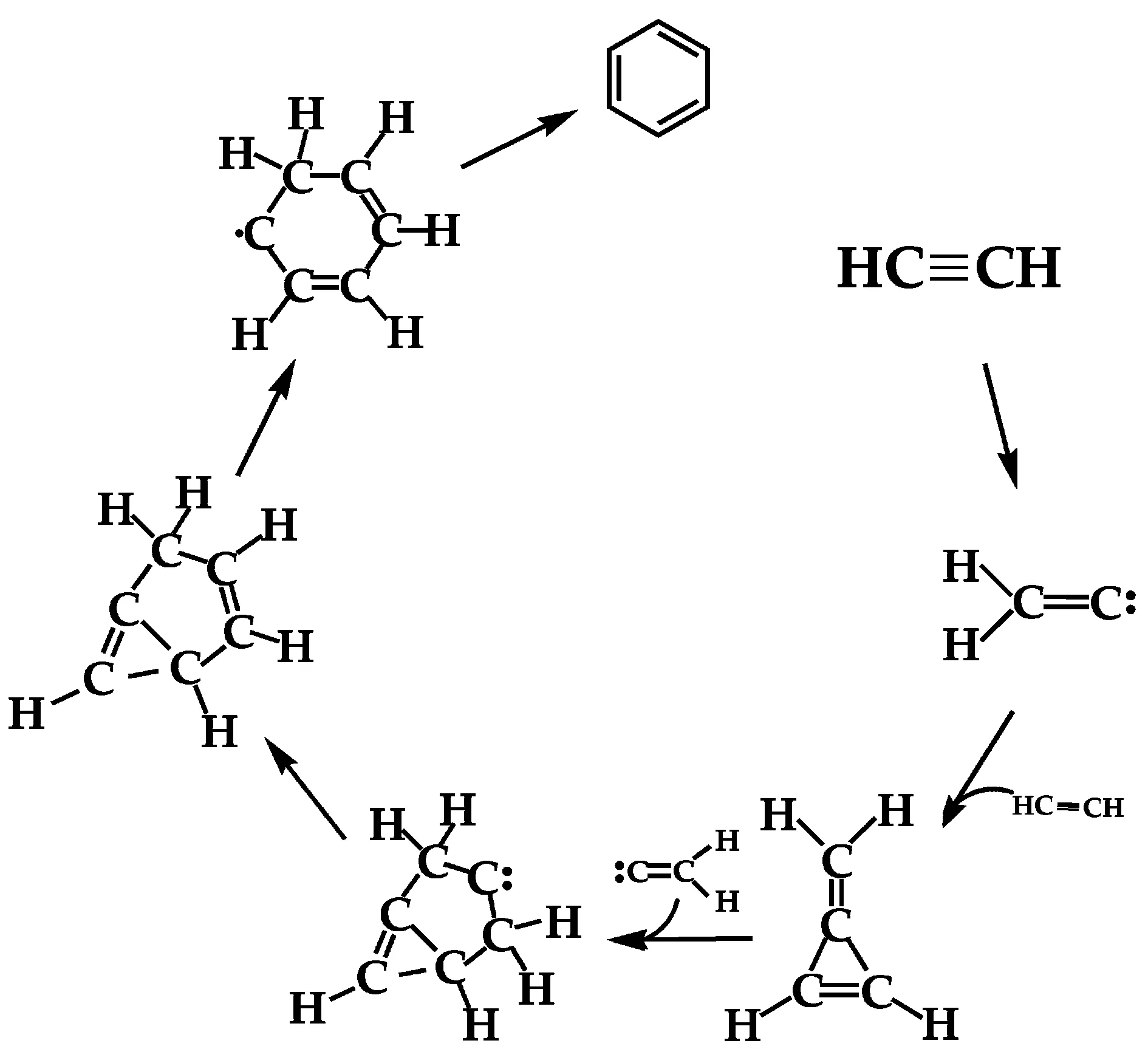
Scheme 1 A brief scheme of the newly proposed reaction pathway.
Though many studies regarding the formation of the aromatic ring have been reported,including the C3H3radical path,C4H4radical path described previously,the premier purpose of the present manuscript is to explore the reaction route via a more general way,i.e.through selecting the products with a larger concentration in the realistic combustion system as the reactants of the present work.Experimentally,acetylene,which has a high emission concentration in internal combustion engines[23–26],is believed to be a key precursor of PAHs[23,26–32].In addition,the acetylene to benzene reaction at high temperatures is itself an important total synthesis reaction in organic chemistry[14]and has been addressed by many studies,including the pioneering work[33–35]by Hopf et al.And a possible mechanism by which benzene is generated from acetylene has been proposed[14].Thus,we select acetylene as the basis to investigate the formation of the aromatic ring using first principles plane-wave based Car-Parrinello molecular dynamics(CPMD)simulations[36].The BLYP exchange-correlation function was used in the CPMD calculations.The ab initio method has been used in various systems[37–41].Electronic structure calculations were performed using the composite method CBS-QB3.Other computational details are provided in supplementary materials.
II.THEORETICAL METHODS
The BLYP exchange-correlation function was used in the CPMD calculations[42–44].The ab initio molecular dynamics were performed using a time step of 4.5 a.u.and a fictitious electron mass of 600 a.u.The energy cuto ffof the plane-wave basis set to expand the electronic wave functions was carefully chosen and was set to 90 Ry(see FIG.S1,Table S1,Table S2,and Table S3 in supplementary materials).Additionally,the Troullier-Martins norm-conserving pseudopotential[45]was used.The temperature of both the ions and electrons in the simulations was controlled using Nose-Hoover thermostats[46–48].The ion temperature was set to 3000 K to accelerate the reactions[49].We note that this temperature is within the experimental range[50].The electronic degrees of freedom were coupled by a coupling frequency of 10000 cm−1[49],and the hydrogen mass was substituted by the deuterium mass[51,52].A total of 20 acetylene molecules were placed in a cubic box with a length of 16˚A,as depicted in FIG.S2.The density of the system is 0.21 g/cm3and is an order of magnitude higher than that reported in experiment[53]to reduce the computational cost because,even with the state of the art computational resources,the ab initio molecular dynamics in gas phase using the exact experimental conditions are hardly manageable for more than a few picoseconds.In addition,the carbene described herein is readily generated through the highly reversible reaction from each other[54,55]and the C2H2molecule is abundant[23–26].Thus,to the best of our knowledge,the results shown here are credible.Periodic boundary conditions and an NVT ensemble were used in this work.Approximately 21.77 ps in this work are required to form a benzene ring,and data within this time period were collected and analyzed.In addition,unless otherwise specified,the geometry optimization and the Gibbs free energy calculations were performed using the composite method CBS-QB3.This method has been shown to be suitable for the calculation of C/H systems[44,56–60].All frequencies were confirmed by way of frequency analyses,and IRC calculations were performed for the transition states to ensure that they corresponded to the intended reactants/products.The molecular dynamics simulations were conducted using the cpmd-3.17.1 code[61],and all electronic structure calculations were performed using the Gaussian 09 software package[62].
III.RESULTS AND DISCUSSION
Unexpectedly,no molecular collisions during the simulated time period induced C−H bond or C−C bond dissociation in acetylene.Instead,the collisions only caused geometric distortions and translations of the acetylene molecules(see supplementary materials for further discussion).This mechanism is plausible given the weak steric hindrance of deformation and the smallmolecule-aided accessible translation of acetylene.The reaction begins with an isomerization of acetylene and the vinylidene carbene,which is consistent with previous reports[63–65]. Given the prominent role of the vinylidene carbene in the newly proposed pathway(see below),the lifetime of this carbene was also determined(FIG.S7(a)and(b)).The results showed that the most vinylidene carbenes survived for more than 100 fs.The shortest lifetime was 38.97 fs and the longest lifetime was 1009.26 fs which is constent with another study[55].Notably,based on the notion that whether the two migrated H atoms are identical dur-ing the formation and reaction of vinylidene,the isomerization of the vinylidene carbene to acetylene was found to have two distinct mechanisms:an H-exchange mechanism(FIG.S7(c))and a non-H-exchange mechanism(FIG.S7(d)).In addition,the conversion between acetylene and vinylidene carbene is so highly reversible from each other[55]that makes the carbene seem to have lived for 3.5µs in the CEI experiment[54].
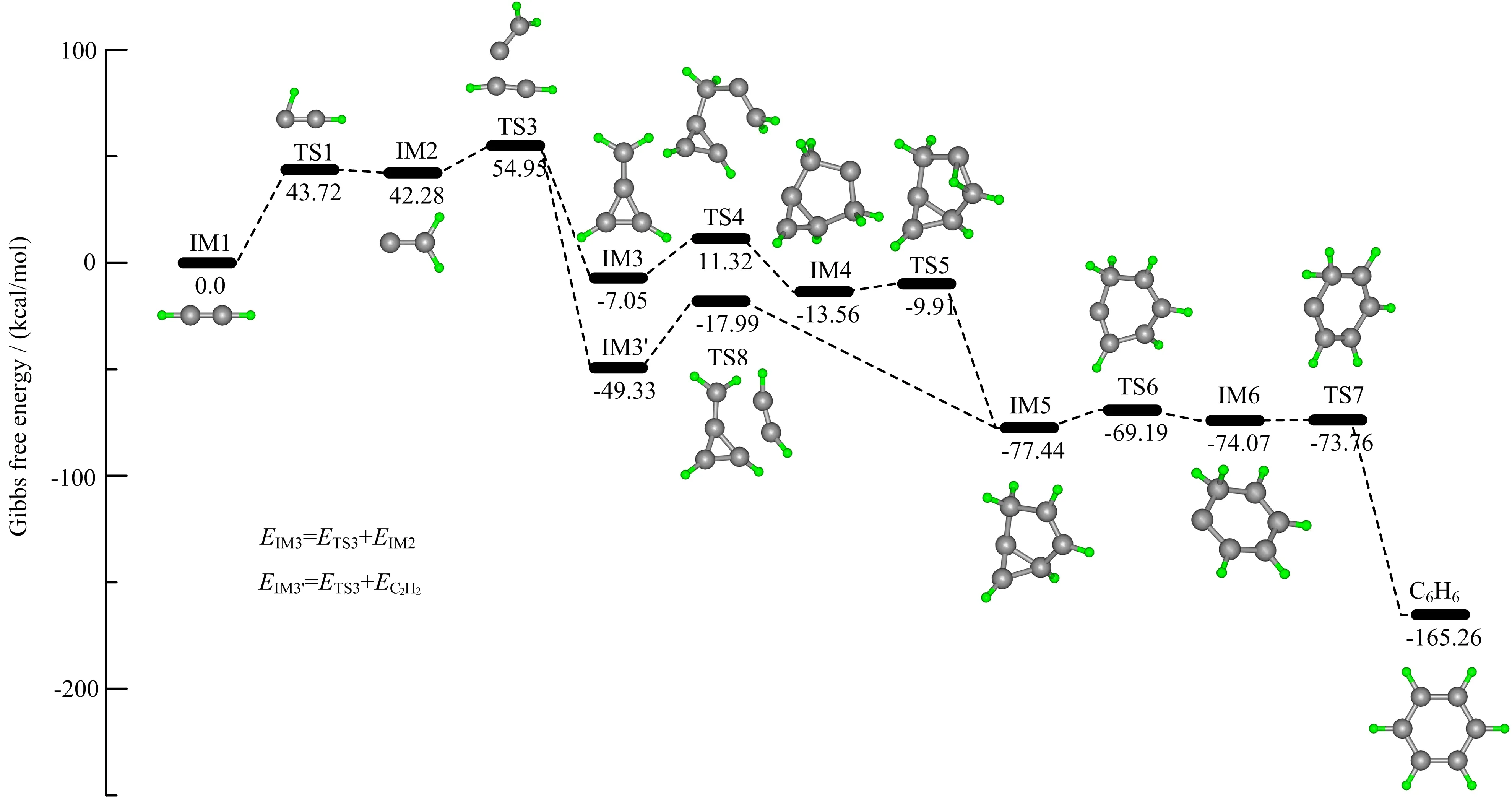
FIG.1 Gibbs free energy diagram for the formation of benzene through successive vinylidene additions.The Gibbs free energy was calculated at 298.15 K,1 atm.Optimized geometries of all intermediates and transition states are also shown.See FIG.S8 for enlarged images of the intermediates and transition states.
We now focus on the formation of the benzene molecule.A brief route is depicted in Scheme 1.Although CPMD simulations can provide an accurate trajectory for the reaction pathway,additional Gibbs free energy changes were calculated at 298.15 K,1 atm to provide a precise energy diagram for quantitatively evaluating the competitiveness of the reaction pathway.The results are depicted in FIG.1.
First,the carbon atom with two unpaired electrons in the vinylidene carbene,which isomerized from acetylene,attacks one acetylene molecule to form methylenecyclopropene(abbreviated as IM3 hereafter).IM3 is then attacked by the vinylidene carbene to eventually form benzene(a video of selected key snapshots is provided for clarity).As seen in FIG.1,acetylene isomerizes to form vinylidene(IM2)through TS1.This process,along with the calculation of rate constants,has been widely discussed and interested readers can refer elsewhere[66–73].Though the calculation of rate constants is not the focus of the present manuscript,it is noteworthy to clarify that one should also consider the concentrations of the reactants besides the rate constants to evaluate the contribution of one reaction route.As a high-energy carbene,vinylidene with the two lone electrons readily pairs with two p-orbital electrons from the two C atoms in acetylene to form IM3 through transition state TS3 with a barrier of 12.68 kcal/mol.And an acetylene π-bond must be broken for this reaction to proceed.In FIG.1,IM3 and IM3′are identical structures,but their relative energies difler based on their respective pathways.IM3 could react with another vinylidene carbene through a 18.37 kcal/mol barrier at transition state TS4 to give IM4,a six C-based ring with its two meta-position C atoms bonded together.FIG.S5(supplementary materials)indicates that IM4 is formed ahead of the H migration to form IM5,which proceeds through TS5 via a 3.65 kcal/mol barrier.Next,the IM5 intermediate proceeds through the meta-position C−C bond dissociation and another intra-molecular H-atom migration with respective barriers of 8.25 and 0.31 kcal/mol to finally generate benzene.In summary,reaction(5)proceeds through roughly four steps:formation of a 5-member ring,hydrogen migration,ring expansion,and another H migration.

FIG.2(a)Acetylene to benzene reaction pathway as described in chapter 5 of Ref.[14].(b)Comparison of the key reaction steps in the two mechanisms.The Gibbs free energy was calculated at 298.15 K,1 atm.
A notable mechanism in Ref.[14],as indicated in FIG.2(a),argues that vinylidene(B)could insert into the C−H bond of acetylene to form vinylacetylene(C).Then,the H atom on the terminal carbon atom of(C)migrates to form(D).The carbon atom with two free electrons in(D)could then insert into the C−H bond of another acetylene to give hexa-1,3-dien-5-yne(E),followed by the isomerization of(E)to form benzene.This route represents a competing pathway to that of our newly proposed mechanism.The key step in the two mechanisms involves whether the vinylidene carbene reacts with acetylene to form methylenecyclopropene(i.e.,from IM2 to IM3 in FIG.1)or is inserted into the C−H bond of acetylene to form vinylacetylene(i.e.,from(B)to(C)in FIG.2(a).To clarify the competitiveness of the two steps in each mechanism,additional calculations were performed.For the reaction from(B)to(C)depicted in FIG.2(a),a transition structure TS9(FIG.2(b))is found.The energy barrier of TS9 is 29.29 kcal/mol,whereas the barrier of the competing step in the newly proposed mechanism is 12.68 kcal/mol,which suggests that the newly proposed pathway is more advantageous than that proposed in Ref.[14]. Additionally,we note that the vinylacetylene carbene cation could isomerize to methylenecyclopropene following exposure to light[74].Kiefer et al.also reported pathways through the IM3 structure,but a cycle opening of IM3,which might make this route less favor because a C=C=C linear bend structure is generated due to the ring opening,is involved in their following route[75,76].
As stated previously,no collisions to induce bond dissociation in acetylene were observed. A single C−H bond dissociation must absorb approximately 131.30 kcal/mol,and approximately 230.60 kcal/mol is required to break a C≡C bond[77].Thus,the first vinylidene reaction with acetylene,which possesses an energy barrier of 12.68 kcal/mol,is preferred.We note that 43.72 kcal/mol in forming vinylidene carbene is the highest energy barrier of the entire pathway.It has also been argued[63–65]that the pyrolysis of acetylene actually begins with its isomerization and that multiple re-crossings between acetylene and vinylidene are realistic because of the coupling between kinetic energy and the vibrational mode[55].The multiple re-crossings are consistent with Coulomb explosion imaging(CEI)experiment[54],in which experiment Levin et al.reported that half the molecules were roughly “still” vinylidene at 3.5µs after the birth of this carbene.Another reaction that might be considered is the addition of a second vinylidene rather than an acetylene molecule.Based on the PES,we see that the energy barriers of adding an acetylene molecule and vinylidene carbene are 31.34 and 18.37 kcal/mol,respectively.Thus,the second vinylidene is preferred.Additionally,reactions involving carbenes are typically faster than those involving closed-shell species.The new route is also supported by results showing that acetylene has a higher emission concentration and is widely believed to be an important contributor to the formation of soot[23,27–29,31,32].Thus,the successive vinylidene addition pathway may be more important in the formation of benzene compared to previously described pathways.
IV.CONCLUSION
This report presents compelling evidence of a viable pathway by which the aromatic ring forms under high temperature conditions.According to the ab initio calculations,benzene can be formed through successive vinylidene additions.No collisions involving acetylene were found to induce C−H bond or C−C bond dissociation.These findings are in direct contrast to previous results suggesting that the formation of benzene molecule occurs through reactions between a C4Hxradical and a C2Hyradical,which are formed through acetylene collisional reactions or between C3 radicals.The reaction pathway presented herein is energetically preferable than the pathway described in[14].Considering the abundance of acetylene,the conversion between acetylene and vinylidene carbene is highly reversible from each other[55],the experimental result that half molecules are “still” vinylidene after 3.5 µs in CEI experiment[54],and the faster reaction of the car-benes involved in the reactions,successive vinylidene additions may contribute significantly to the formation of benzene under certain conditions.In addition,most of the vinylidene carbenes in the present simulation were found to survive for more than 100 fs.Consequently,we expect this mechanism to be helpful in clarifying the formation of aromatic compounds.At last,these theoretical results await direct comparison from experiments,which hopefully could be provided by time-resolved coulomb explosion imaging experiments[78].
Supplementary materials:Further discussions on the eflects brought by molecular collisions and definitions of lifetime of vinylidene are provided in the supporting materials.We provide also additional information about validations of CPMD calculations,enlarged images and optimized coordinates of intermediates involved in the newly proposed reaction route.
V.ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China(No.21403221 and No.91441106)
[1]D.M.Whitacre,Reviews of Environmental Contamination and Toxicology,New York:Springer,(2010).
[2]E.J.Barrientos,M.Lapuerta,and A.L.Boehman,Combust.Flame 160,1484(2013).
[3]M.Berthelot,Ann.Chem.Pharmacie 141,177(1867).
[4]J.D.Bittner and J.B.Howard,Symp.(Int.)Combust.18,1105(1981).
[5]M.Frenklach,D.W.Clary,W.C.Gardiner Jr.,and S.E.Stein,Symp.(Int.)Combust.20,887(1985).
[6]M.Frenklach and H.Wang,Symp.(Int.)Combust.23,1559(1991).
[7]D.S.Parker,R.I.Kaiser,T.P.Troy,and M.Ahmed,Angew.Chem.Int.Ed.Engl.53,7740(2014).
[8]V.V.Kislov,A.I.Sadovnikov,and A.M.Mebel,J.Phys.Chem.A 117,4794(2013).
[9]A.M.Mebel,V.V.Kislov,and R.I.Kaiser,J.Am.Chem.Soc.130,13618(2008).
[10]K.Ono,Y.Matsukawa,K.Dewa,A.Watanabe,K.Takahashi,Y.Saito,Y.Matsushita,H.Aoki,K.Era,T.Aoki,and T.Yamaguchi,Combust.Flame 162,2670(2015).
[11]B.Shukla and M.Koshi,Phys.Chem.Chem.Phys.12,2427(2010).
[12]B.Shukla and M.Koshi,Combust.Flame 159,3589(2012).
[13]P.R.Westmoreland,A.M.Dean,J.B.Howard,and J.P.Longwell,J.Phys.Chem.93,8171(1989).
[14]H.Hopf,Modern Arene Chemistry,D.Astruc Ed.,Weinheim:Wiley-VCH,(2002).
[15]M.Frenklach,Phys.Chem.Chem.Phys.4,2028(2002).
[16]M.Frenklach,W.C.Gardiner,S.E.Stein,D.W.Clary,and T.Yuan,Combust.Sci.Technol.50,79(1986).
[17]M.Frenklach,D.W.Clary,W.C.Jr.Gardiner,and S.E.Stein,Symp.(Int.)Combust.21,1067,(1988).
[18]M.Frenklach,T.Yuan,and M.Ramachandra,Energy Fuels 2,462(1988).
[19]C.Wu and R.D.Kern,J.Phys.Chem.91,6291(1987).
[20]U.Alkemade and K.Z.Homann,Z.Phys.Chem.161,19(1989).
[21]S.E.Stein,J.A.Walker,M.M.Suryan,and A.Fahr,Symp.(Int.)Combust.23,85,(1991).
[22]J.A.Miller and C.F.Melius,Combust.Flame 91,21(1992).
[23]S.J.Harris and A.M.Weiner,Annu.Rev.Phys.Chem.36,31(1985).
[24]D.R.Tree and K.I.Svensson,Prog.Energy Combust.Sci.33,272(2007).
[25]D.D.Parrish,W.C.Kuster,M.Shao,Y.Yokouchi,Y.Kondo,P.D.Goldan,de J.A.Gouw,M.Koike,and T.Shirai,Atmos.Environ.43,6435(2009).
[26]V.Chernov,M.J.Thomson,S.B.Dworkin,N.A.Slavinskaya,and U.Riedel,Combust.Flame 161,592(2014).
[27]R.N.Pease,J.Am.Chem.Soc.51,3470(1929).
[28]S.Macadam,J.M.Be´er,A.F.Safofim,and A.B.Ho ffmann,Symp.(Int.)Combust.26,2295(1996).
[29]H.B¨ohm and H.Jander,Phys.Chem.Chem.Phys.1,3775(1999).
[30]Q.Y.Feng,A.Jalali,A.M.Fincham,Y.Lee Wang,T.T.Tsotsis,and F.N.Egolfopoulos,Combust.Flame 159,1876(2012).
[31]L.Figura,F.Carbone,and A.Gomez,Proc.Combust.Inst.35,1871(2015).
[32]H.Omidvarborna,A.Kumar,and D.S.Kim,Renew.Sustain.Energy Rev.48,635(2015).
[33]H.Hopf and H.Musso,Angew.Chem.Int.Ed.Engl.8,680(1969).
[34]H.Hopf,H.Berger,G.Zimmermann,U.Nchter,P.G.Jones,and I.Dix,Angew.Chem.Int.Ed.Engl.36,1187(1997).
[35]M.Prall,A.Kr¨uger,P.R.Schreiner,and H.Hopf,Chemistry 7,4386(2001).
[36]R.Car and M.Parrinello,Phys.Rev.Lett.55,2471(1985).
[37]S.F.Wang and X.J.Wu,Chin.J.Chem.Phys.28,588(2015).
[38]X.H.Lu,D.S.Wang,and J.J.Ming,Chin.J.Chem.Phys.29,193(2016).
[39]W.X.Pang,Y.B.Sun,J.J.Zhao,and Y.Lu,Chin.J.Chem.Phys.29,657(2016).
[40]D.Z.Li,Z.F.Chen,Z.J.Zhang,and J.Liu,Chin.J.Chem.Phys.30,735(2017).
[41]X.X.Zhao,H.Q.Chen,and B.Li,Chin.J.Chem.Phys.30,529(2017).
[42]C.Lee,W.Yang,and R.G.Parr,Phys.Rev.B 37,785(1988).
[43]A.D.Becke,J.Chem.Phys.98,5648(1993).
[44]J.A.Jr.Montgomery,M.J.Frisch,J.W.Ochterski,and G.A.Petersson,J.Chem.Phys.110,2822(1999).
[45]N.Troullier and J.L.Martins,Phys.Rev.B 43,1993(1991).
[46]W.G.Hoover,Phys.Rev.A 31,1695(1985).
[47]S.Nos´e,Mol.Phys.52,255(1984).
[48]S.Nos´e,J.Chem.Phys.81,511(1984).
[49]M.S.Mettler,S.H.Mushrif,A.D.Paulsen,A.D.Javadekar,D.G.Vlachos,and P.J.Dauenhauer,Energy Environ.Sci.5,5414(2012).
[50]Y.Yoshizawa,H.Kawada,and M.Kurokawa,Symp.(Int.)Combust.17,1375(1979).
[51]P.Durlak and Z.Latajka,Phys.Chem.Chem.Phys.16,23026(2014).
[52]S.K.Sahoo and N.N.Nair,J.Comput.Chem.37,1657(2016).
[53]T.Mendiara,M.P.Domene,A.Millera,R.Bilbao,and M.U.Alzueta,J.Anal.Appl.Pyrolysis 74,486(2005).
[54]J.Levin,H.Feldman,A.Baer,D.Ben-Hamu,O.Heber,D.Zajfman,and Z.Vager,Phys.Rev.Lett.81,3347(1998).
[55]R.L.Hayes,E.Fattal,N.Govind,and E.A.Carter,J.Am.Chem.Soc.123,641(2001).
[56]G.Petersson,A.Bennett,T.G.Tensfeldt,M.A.Al-Laham,W.A.Shirley,and J.Mantzaris,J.Chem.Phys.89,2193(1988).
[57]G.Petersson and M.A.Al-Laham,J.Chem.Phys.94,6081(1991).
[58]G.A.Petersson,T.G.Tensfeldt,and J.A.Montgomery Jr.,J.Chem.Phys.94,6091(1991).
[59]J.A.Jr.Montgomery,J.W.Ochterski,and G.A.Petersson,J.Chem.Phys.101,5900(1994).
[60]J.W.Ochterski,G.A.Petersson,and J.A.Montgomery Jr.,J.Chem.Phys.104,2598(1996).
[61]S.P.Walch,J.Chem.Phys.103,8544(1995).
[62]M.J.Frisch,G.W.Trucks,H.B.Schlegel,G.E.Scuseria,M.A.Robb,J.R.Cheeseman,G.Scalmani,V.Barone,B.Mennucci,G.A.Petersson,H.Nakatsuji,M.Caricato,X.Li,H.P.Hratchian,A.F.Izmaylov,J.Bloino,G.Zheng,J.L.Sonnenberg,M.Hada,M.Ehara,K.Toyota,R.Fukuda,J.Hasegawa,M.Ishida,T.Nakajima,Y.Honda,O.Kitao,H.Nakai,T.Vreven,J.A.Montgomery,Jr.,J.E.Peralta,F.Ogliaro,M.Bearpark,J.J.Heyd,E.Brothers,K.N.Kudin,V.N.Staroverov,R.Kobayashi,J.Normand,K.Raghavachari,A.Rendell,J.C.Burant,S.S.Iyengar,J.Tomasi,M.Cossi,N.Rega,J.M.Millam,M.Klene,J.E.Knox,J.B.Cross,V.Bakken,C.Adamo,J.Jaramillo,R.Gomperts,R.E.Stratmann,O.Yazyev,A.J.Austin,R.Cammi,C.Pomelli,J.W.Ochterski,R.L.Martin,K.Morokuma,V.G.Zakrzewski,G.A.Voth,P.Salvador,J.J.Dannenberg,S.Dapprich,A.D.Daniels,.Farkas,J.B.Foresman,J.V.Ortiz,J.Cioslowski,and D.J.Fox,Gaussian 09,Wallingford,CT,USA:Gaussian,Inc.,(2009).
[63]W.Reppe and W.J.Schweckendiek,Justus Liebigs Ann.Chem.560,104(1948).
[64]W.Reppe,Experientia 5,93(1949).
[65]R.P.Duran,V.T.Amorebieta,and A.J.Colussi,J.Phys.Chem.92,636(1988).
[66]C.E.Dykstra and H.F.Schaefer III,J.Am.Chem.Soc.100,1378(1978).
[67]K.A.Peterson and T.H.Jr.Dunning,J.Chem.Phys.106,4119(1997).
[68]R.F.C.Brown,Eur.J.Org.Chem.1999,3211(1999).
[69]T.Osipov,C.L.Cocke,M.H.Prior,A.Landers,T.Weber,O.Jagutzki,L.Schmidt,H.Schmidt-B¨ocking,and R.Drner,Phys.Rev.Lett.90,233002(2003).
[70]S.L.Zou,J.M.Bowman,and A.Brown,J.Chem.Phys.118,10012(2003).
[71]R.F.C.Brown,Pyrolytic Methods in Organic Chemistry:Application of Flow and Flash Vacuum Pyrolytic Techniques,New York:Elsevier,(2012).
[72]H.Lee,J.H.Baraban,R.W.Field,and J.F.Stanton,J.Phys.Chem.A 117,11679(2013).
[73]L.F.Guo,H.X.Han,J.Y.Ma,and H.Guo,J.Phys.Chem.A 119,8488(2015).
[74]G.Koster and W.Van der Hart,Int.J.Mass Spectrom.Ion Processes 163,169(1997).
[75]J.H.Kiefer and W.A.Von Drasek,Int.J.Chem.Kinet.22,747(1990).
[76]J.H.Kiefer,Int.J.Chem.Kinet.25,215(1993).
[77]K.M.Ervin,S.Gronert,S.E.Barlow,M.K.Gilles,A.G.Harrison,V.M.Bierbaum,C.H.DePuy,W.C.Lineberger and G.B.Ellison,J.Am.Chem.Soc.112,5750(1990).
[78]H.Ibrahim,B.Wales,S.Beaulieu,B.E.Schmidt,N.Thir,E.P.Fowe,.Bisson,C.T.Hebeisen,V.Wanie,M.Gigu´ere,Nat.Commun.5,5422(2013).
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Imaging HNCO Photodissociation at 201 nm:State-to-State Correlations between CO(X1Σ+)and NH(a1∆)
- Energy-Transfer Processes of Xe(6p[1/2]0,6p[3/2]2,and 6p[5/2]2)Atoms under the Condition of Ultrahigh Pumped Power
- Ultrafast Investigation of Excited-State Dynamics in Trans-4-methoxyazobenzene Studied by Femtosecond Transient Absorption Spectroscopy
- Strong Current-Polarization and Negative Diflerential Resistance in FeN3-Embedded Armchair Graphene Nanoribbons
- Direct Observation of Transition Metal Dichalcogenides in Liquid with Scanning Tunneling Microscopy
- Photo-Induced Intermolecular Electron Transfer-Eflect of Acceptor Molecular Structures