4-氨基安替比林电子光谱及溶剂效应理论研究
2019-01-09鲁礼林阮祝华倪嘉琪舒红飞
鲁礼林,陈 俊,阮祝华,倪嘉琪,舒红飞
(1.武汉科技大学化学与化工学院,湖北 武汉,430081;2.武汉科技大学煤转化与新型炭材料湖北省重点实验室,湖北 武汉 430081)
吡唑啉酮类杂环化合物是非甾类药物如治疗关节炎药物的主要活性成分[1],而4-氨基安替比林(4-AAP)是其中最常见、应用最广泛的一种衍生物,因其在紫外可见光区具有优良的光吸收特性,故常被用做显色剂来制备染料和颜料,或作为光分析试剂对水体环境中一些有害物质如二氧化氯、酚类化合物、残杀威及金属离子如Zn2+、Al3+、Cr3+和砷酸盐等进行检测[2-7],此外它在药物分析与生化检验领域可用于测定羟甲叔丁肾上腺素、羟苄羟麻黄碱等的含量[8-9],构建针对葡萄糖、乳酸盐、乙醇、半乳糖和氨基酸含量进行分析的双酶分析微反应器[10],检测维生素B6注射液中维生素B6的含量[11]。虽然4-AAP在光学分析方面应用广泛,但仍有必要对其电子光谱特性进行深入研究,以便进一步了解其光谱吸收性质及明确相关影响因素,更准确地归属其吸收谱带。
借助量子化学计算,根据电子跃迁轨道拓扑结构对化合物电子光谱谱带进行更准确的归属,是目前研究电子吸收光谱特性的重要手段,而理论计算模型的正确选择则是其中关键之所在。早期的Pariser-Parr-Pople(PPP)法和ZINDO/S法都是典型的半经验研究方法,此类方法适用范围有限,只能针对一些特定体系取得较精确结果。当前应用最为广泛的是基于Hartree-Fock的单激发组态相互作用(CIS)方法和随机相近似方法(RPA),但由于其忽略了电子相关作用,导致在很多体系中计算结果偏差较大。此外,耦合簇参考态运动方程(EOM-CC)、多参考态组态相互作用(MRCI)以及包含二级微扰的多组态自洽场(CASSCF/CASPT2)等高度电子相关方法虽在计算激发能方面表现优异,但它们计算成本高昂,目前仅限于较小体系的模拟计算,且难以考虑溶剂化效应对激发能的影响。而含时密度泛函方法(TDDFT)通过对时间和空间进行数值积分来求解薛定谔方程,同时可加入显性或隐性溶剂化模型考虑溶剂效应,且计算精度与高度电子相关方法相当,是目前研究化合物和金属配合物电子结构、激发光谱及非线性光学性质较理想的一种方法。故本文使用PCM-TDPBE0/6-311++G(d, p)//PBE0/6-311G(d, p)密度泛函方法,结合分子动力学模拟及紫外光谱试验,研究了4-AAP分子及其在水溶液中与溶剂水分子的氢键复合物结构、电子光谱以及溶剂环境、水溶液中溶剂水分子与4-AAP分子氢键相互作用对于电子光谱特性的影响。
1 实验与计算方法
1.1 实验
利用UV-2550岛津紫外可见光谱仪在乙醇和水溶液两种环境中分别测定4-AAP的UV-Vis吸收光谱,所用试剂均为分析纯。
1.2 计算方法
4-AAP分子及其氢键复合物结构搜索优化均使用PBE0杂化泛函[12]和6-311G(d, p)基组,利用简振频率计算确定相关结构的稳定性,使用含时密度泛函 TD-PBE0[13]和6-311++G(d, p)基组计算4-AAP分子及其氢键复合物的激发跃迁光谱,通过极化连续模型(PCM)考虑溶剂效应对激发能的影响,使用经过零点能校正的电子能量进行氢键复合能计算,所有密度泛函计算工作使用Gaussian09[14]完成。
水溶液中4-AAP分子与溶剂水分子相互作用的团簇模型研究采用经典分子动力学方法,使用Materials Studio 4.3 软件包中的Discover模块优化结构和正则系综(NVT)分子动力学模拟。模拟温度为298 K, Anderson法控温,使用COMPASS力场参数,首先构建内含256个溶剂分子和1个4-AAP分子的周期性边界体系(a=b=c=19.67 Å,α=β=γ=90°),经100 ps预平衡后进行5 ns时长动力学模拟平衡,步长为1 fs,运用Velocity-Verlet法求解牛顿运动方程。
2 结果与讨论
2.1 4-AAP的UV-Vis实验光谱
4-AAP在乙醇和水溶液两种环境中的UV-Vis吸收光谱测试结果如图1所示。由图1可见,4-AAP在紫外可见光区230~260 nm存在最强吸收峰,270~300 nm处呈现吸光度稍弱的肩峰,其中水溶液环境下4-AAP的UV-Vis吸收光谱中吸收峰波长位置相对于其在乙醇环境下的相应位置向短波长方向发生大约16nm的蓝移,表明溶剂环境对4-AAP的UV-Vis吸收光谱影响显著。
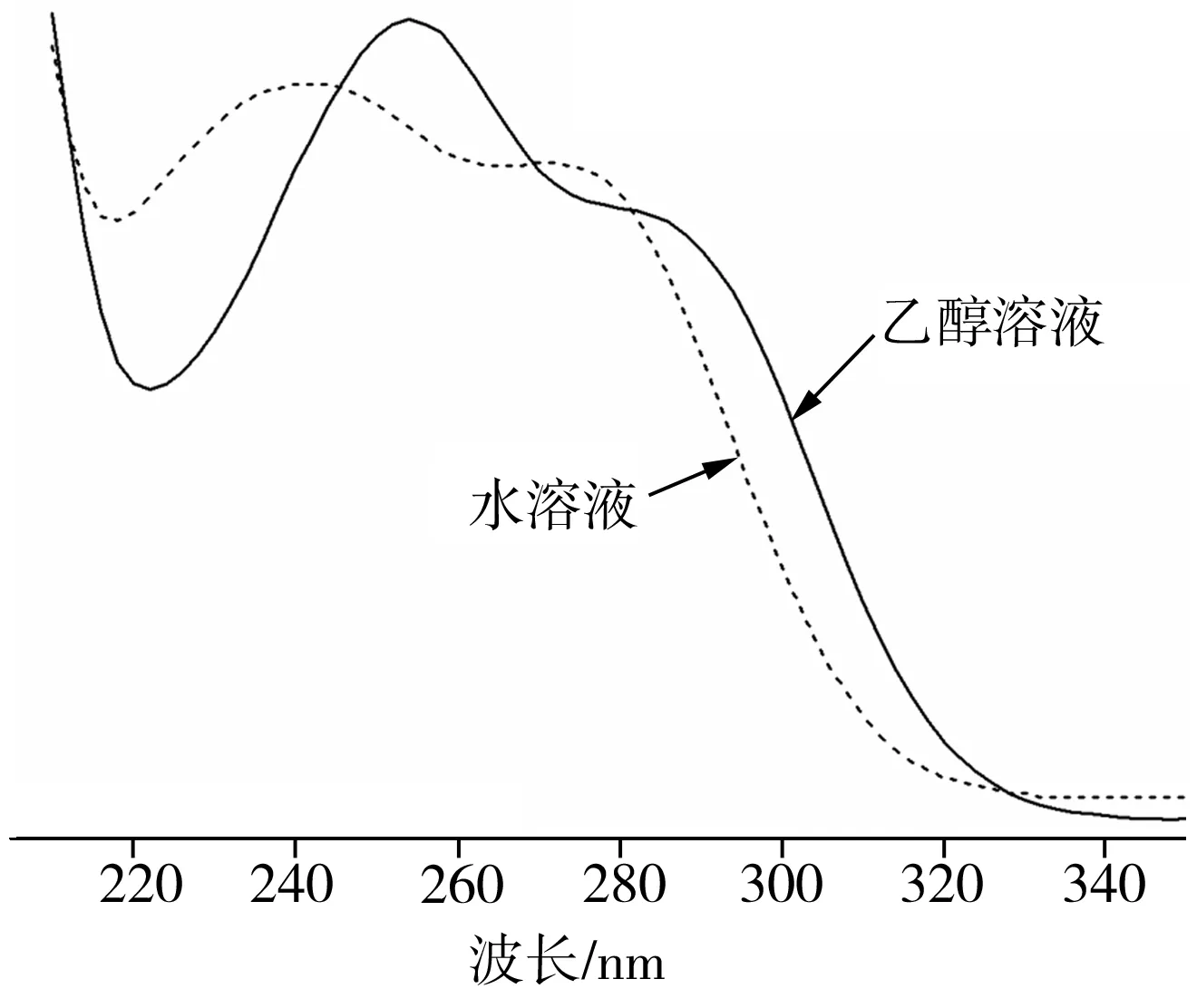
图1 4-AAP的UV-Vis吸收光谱Fig.1 UV-Vis absorption spectra of 4-AAP
2.2 4-AAP分子结构及电子光谱
4-AAP分子结构及原子标识如图2所示,使用密度泛函理论方法PBE0/6-311G(d, p)优化所得稳定的4-AAP基态分子结构参数列于表1中,通过与实验测定的分子结构参数[15]对比分析可知,PBE0杂化泛函能较准确地对4-AAP的分子结构进行描述,最大键长偏差不超过0.03 Å,与B3LYP/6-311G(d,p)和B3LYP/cc-PVDZ计算结果[16]精度相当,这表明利用密度泛函计算模型PBE0/6-311G(d, p)研究4-AAP分子结构可获得满意的计算精度。在电子光谱模拟计算方面PBE0杂化密度泛函明显优于杂化泛函B3LYP而被广泛使用[17],因此本文选用PBE0泛函对4-AAP的分子结构、团簇结构实施优化并进行电子光谱计算。
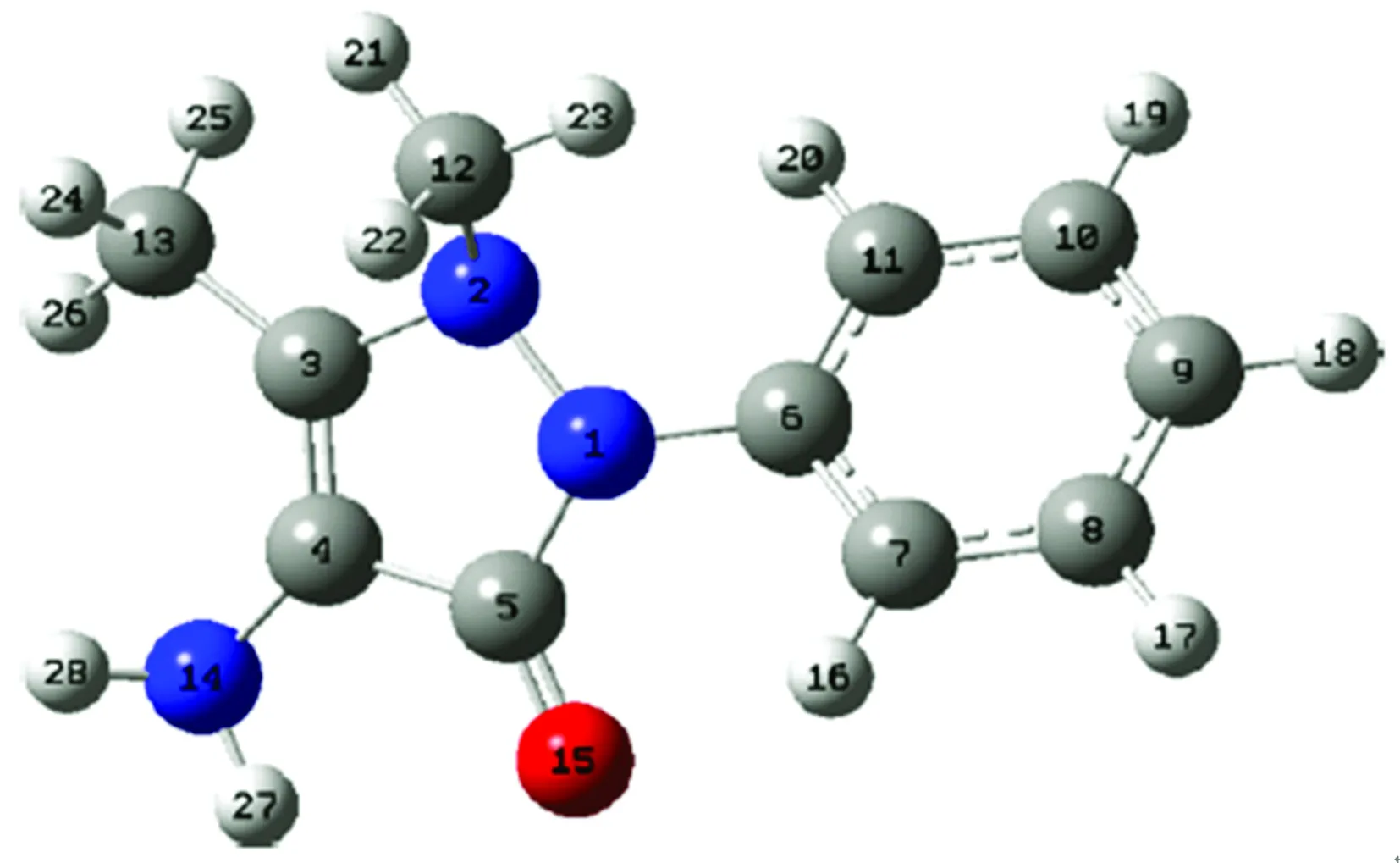
图2 4-AAP分子结构及原子标识Fig.2 Molecular structure of 4-AAP with atomic labels
4-AAP在乙醇溶液和水溶液环境中的模拟电子光谱吸收带及电子跃迁贡献列于表2,其与电子跃迁相关分子轨道的拓扑结构如图3所示。从表2可以看出,在乙醇溶液中安替比林位于259 nm电子光谱最强吸收带,对应于HOMO-1→LUMO轨道电子跃迁,具有典型的π→π*跃迁特征。255 nm处较强的吸收带对应电子HOMO→LUMO+2轨道跃迁,具有电荷转移跃迁特征。HOMO→LUMO轨道电子跃迁对应的电子光谱吸收带吸收波长为290 nm,由分子轨道拓扑结构图可看出从吡唑五元杂环向苯环发生了电荷转移跃迁。
由表2还可以看出,使用PCM模型计算所得4-AAP在乙醇溶液中的最强特征吸收带波长位置与其在乙醇溶液中实验测定的相应值基本一致,但计算所得水溶液中4-AAP理论吸收谱带波长同其实验测定的相应值相差较大,这可能是因为水分子与乙醇分子相比具有更强的极性,前者与4-AAP分子能形成更强的氢键作用,改变了其分子轨道能级及电子垂直激发能从而显现更强的溶剂效应,文献[18-19]的研究也表明,溶剂分子与溶质分子间通过氢键作用形成了团簇复合物,可对其电子吸收光谱产生明显的影响。

表1 4-AAP分子结构键长、键角及二面角参数Table 1 Bond lengths, angle and dihedral angle in 4-AAP molecular structure
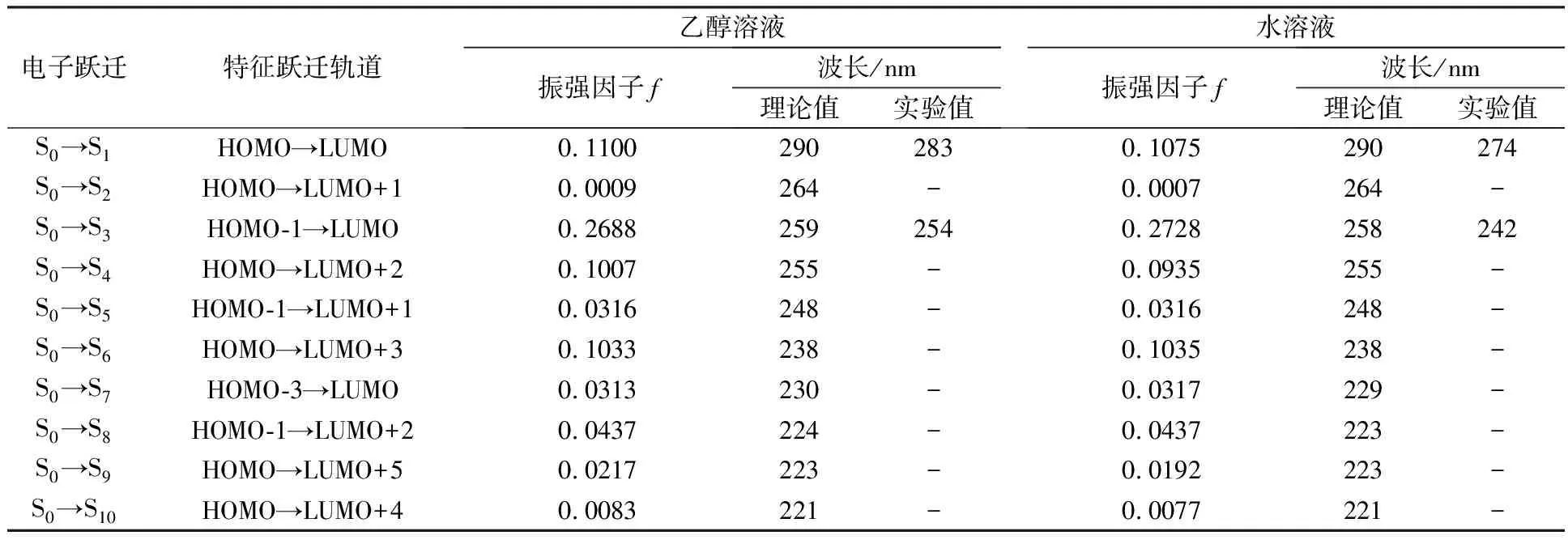
表2 4-AAP电子光谱吸收带及电子跃迁轨道贡献Table 2 Absorption bands in 4-AAP electronic spectra and electron transition involved
2.3 4-AAP水溶液中氢键复合物结构及电子光谱
4-AAP分子与水分子形成的氢键复合物结构如图4所示,表3列出了各氢键复合物中4-AAP分子与水分子的氢键结合能及电子吸收光谱中最强特征吸收带波长位置。由表3可知,氢键复合物结构及氢键形成模式对于4-AAP分子电子光谱吸收带波长位置产生了显著的影响。4-AAP分子中氨基N原子可与水分子中的H原子通过强氢键作用形成复合物结构(见图4(a)),使得肩峰吸收波长位置发生明显蓝移(由290 nm移至276 nm)。通过羰基O与水分子中H原子间氢键形成复合物(见图4(b))对肩峰几乎无影响,但能使最强吸收峰位置发生较明显的蓝移(由258 nm移至254 nm)。当水分子同时与4-AAP分子中羰基O和氨基H原子形成氢键时(见图4(c)),更高的氢键结合能使得复合物更加稳定,最强吸收峰波长基本不变,但肩峰波长位置发生明显红移(由290 nm红移至298 nm)。通过吡唑杂环上甲基相连的N原子与水分子形成氢键(见图4(d)),最强吸收峰基本不受影响,却使肩峰波长位置发生较明显蓝移。4-AAP分子通过氨基H原子与水分子形成复合物(见图4(e)),导致肩峰波长发生明显红移。当两个水分子同时与4-AAP分子中羰基O和氨基形成氢键时,根据水分子与氨基形成氢键方式的不同可得两种复合物结构分别如图4(f)、4(g)所示,其中以N…H-O氢键结合比以N-H…O结合物更稳定。由表3可知,两种成键模式对于最强吸收峰影响并不明显,但可使肩峰位置发生不同的位移,水分子与氨基N原子形成氢键(N…H-O)导致吸收峰位置发生蓝移,而与氨基H原子形成氢键(N-H…O)则导致吸收峰位置明显红移。与羰基O形成不同数量氢键的复合物结构分别如图4(h)、图4(i)所示,综合表3结果可知,最强吸收峰波长位置受氢键数量影响不明显。一个水分子同时与氨基H原子和羰基O原子形成氢键,使得肩峰波长位置发生约10nm的红移,但与羰基O形成氢键的水分子数量对其影响不大。当水分子数量增加至4时,第4个水分子由于空间位阻难于与4-AAP直接形成氢键,而是在水分子间形成氢键(见图4(j))。由以上分析可知,由于溶剂水分子能与4-AAP分子通过分子间氢键作用形成复合物,可对4-AAP电子光谱中主要吸收带产生明显的影响,但不同氢键形成方式的影响作用存在差异,通过羰基O原子形成氢键使得最大吸收峰位置发生蓝移,通过氨基N原子与水分子形成氢键使得肩峰波长位置发生明显蓝移,而通过氨基H原子与水分子形成氢键使得肩峰波长发生明显红移,但对最强吸收峰位置影响不大。通过跃迁轨道分析,所有复合物吸收带电子轨道跃迁贡献与4-AAP分子相同,即最强吸收带对应HOMO-1→LUMO电子轨道跃迁,长波长处吸收带与HOMO→LUMO电荷转移跃迁对应。与实验测定的吸收光谱进行比较,可推测在水溶液中4-AAP分子主要通过羰基O原子和氨基N原子与水分子间的氢键作用形成复合物,从而导致4-AAP分子吸收光谱的最强吸收峰和肩峰波长位置均发生蓝移。

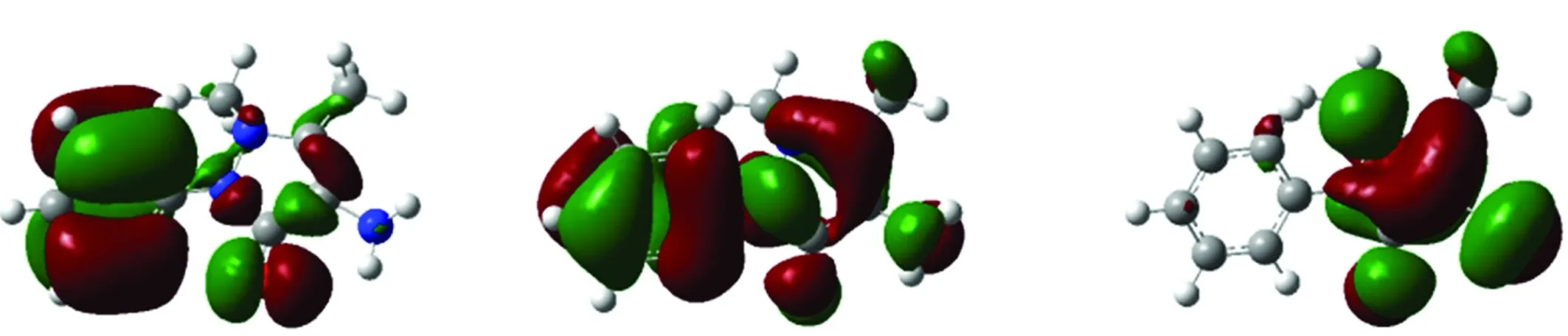
图3 4-AAP分子轨道拓扑结构图及分子轨道能级Fig.3 Sketch and energies of selected molecular orbitals involved in 4-AAP
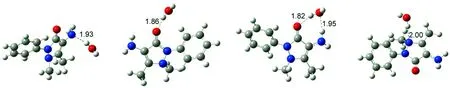
(a)复合物A (b)复合物B (c)复合物C (d)复合物D
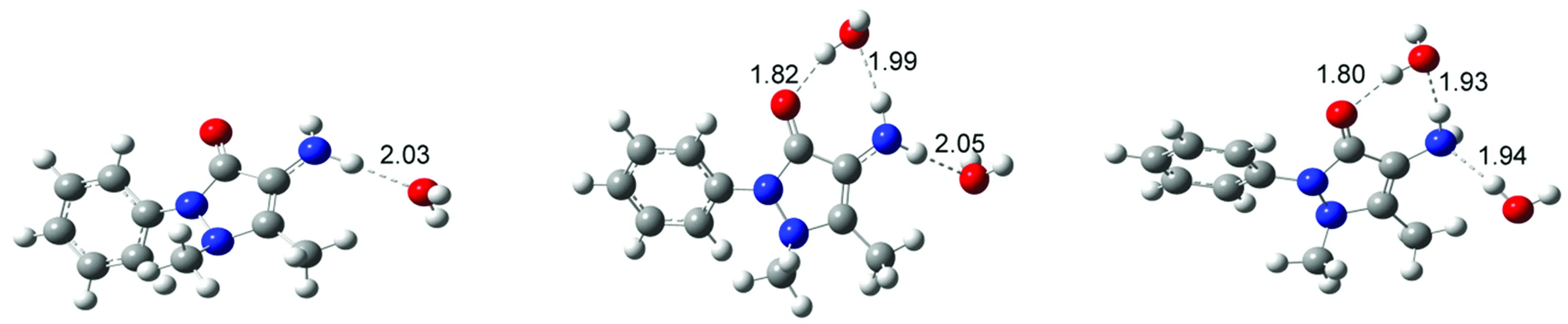
(e)复合物E (f)复合物F (g)复合物G
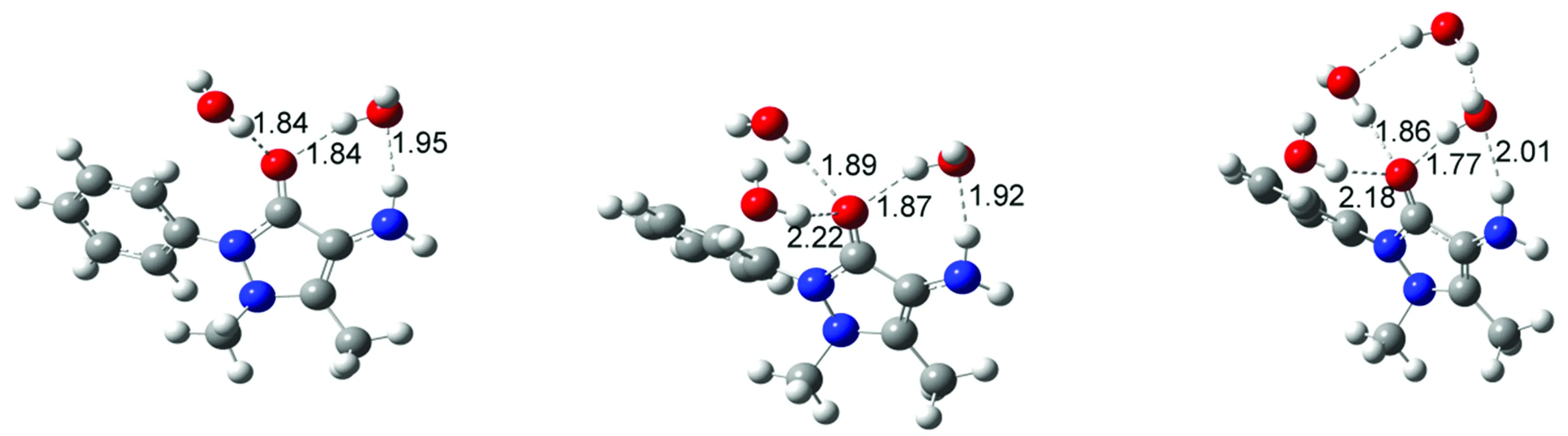
(h)复合物H (i)复合物I (j)复合物J
图44-AAP与水分子氢键复合物结构
Fig.4Structuresofhydrogenbondingcomplexesof4-AAPandH2O
表3氢键复合物结合能及电子光谱吸收带波长
Table3Bindingenergiesofhydrogenbondingincomplexesandwavelengthofabsorptionbandsinelectronicspectra
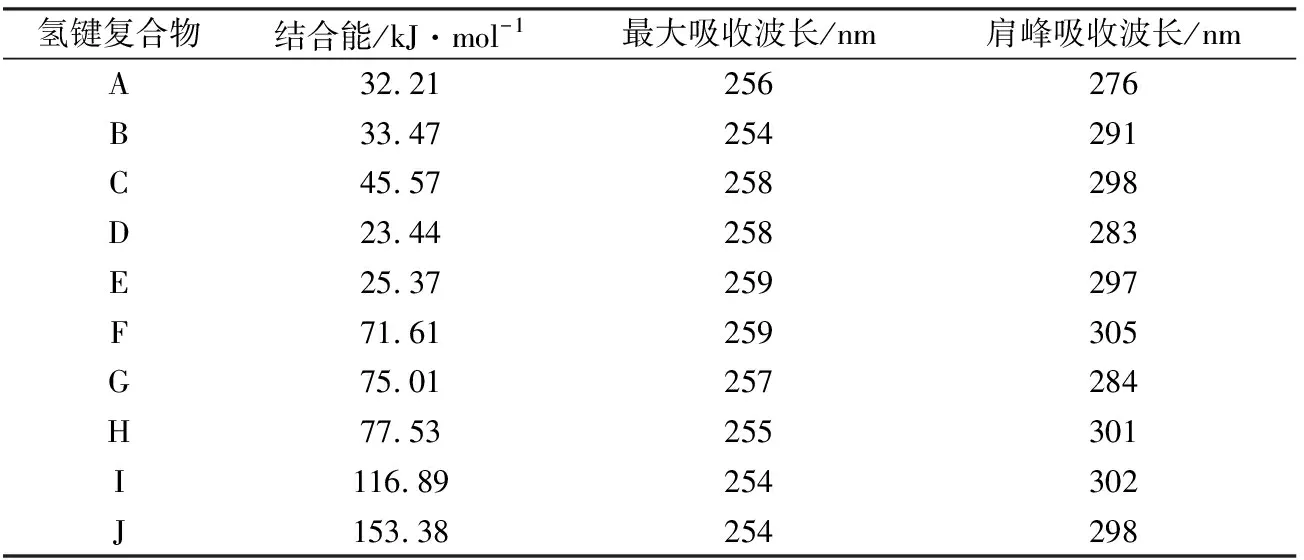
氢键复合物结合能/kJ·mol-1最大吸收波长/nm肩峰吸收波长/nmA32.21256276B33.47254291C45.57258298D23.44258283E25.37259297F71.61259305G75.01257284H77.53255301I116.89254302J153.38254298
2.4 4-AAP水溶液中溶剂分子团簇分子动力学模拟研究
选取COMPASS力场参数进行了分子动力学模拟并通过4-AAP分子中N、O原子与水分子间的径向分布函数分析了氢键形成情况。4-AAP分子中N、O杂原子分别与Hw(水中氢原子)及氨基H原子与Ow(水中氧原子)的径向分布函数如图5所示。由图5(a)可以看出,在径向距离2.0 Å附近杂环N原子(Nmethyl和Nphenyl)与Hw原子之间无径向分布峰出现,这表明4-AAP分子中五元杂环上的N原子难于与水分子间形成氢键作用。但氨基N原子与Hw径向分布图中在径向距离为2.0 Å处出现一个径向分布函数峰,通过对分布峰积分可得N原子与Hw的配位数约为0.70,表明氨基N原子可与水分子形成一个氢键。由图5(b)可见,在4-AAP分子中羰基O原子与Hw间径向分布图中径向距离为2.0 Å处出现较强的径向分布峰,这表明羰基O易与Hw通过氢键作用结合溶剂水分子,积分可知其配位数约为1.87,表明氧原子与两个水分子形成氢键。另外,4-AAP分子中的氨基除了通过N原子与Hw形成氢键外,还可能通过氨基H原子与水分子中的Ow形成氢键。由图5(c)可看出,在距离氨基H原子约3.47 Å出现明显径向函数分布峰,在2.20 Å处分布峰极弱,积分可得氨基H原子与Ow的配位数约为0.22,表明氨基H原子难于与溶剂水分子的Ow间通过氢键作用形成复合物。由分子动力学模拟分析可知,在水溶液中4-AAP分子主要通过羰基O和氨基N原子与水分子氢键结合形成氢键复合物,这与上述光谱分析结论一致,因此可推测出水溶液中4-AAP与水分子形成的氢键复合物结构,其优化后的溶剂团簇模型4-AAP-(H2O)3如图6所示。
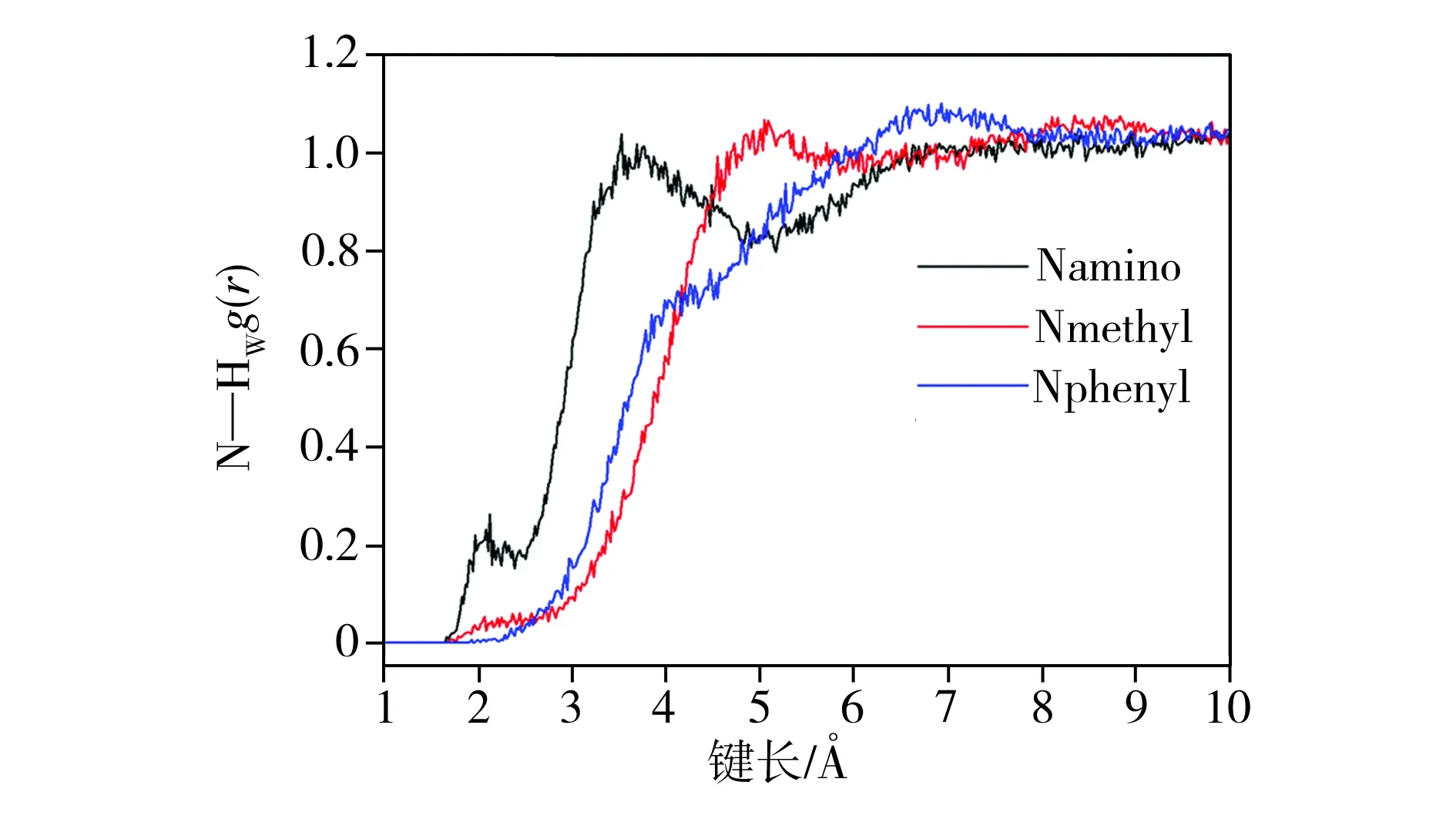
(a)N原子与Hw

(b)羰基O原子与Hw
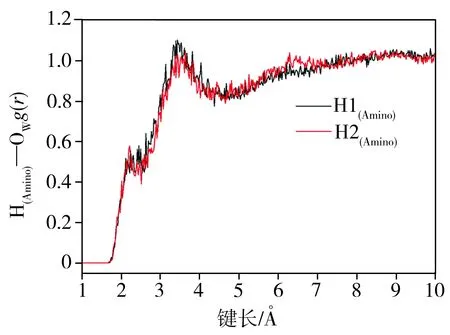
(c)氨基H原子与Ow
Fig.5Radialdistributionfunctions(RDFs)between4-AAPandwatermolecules
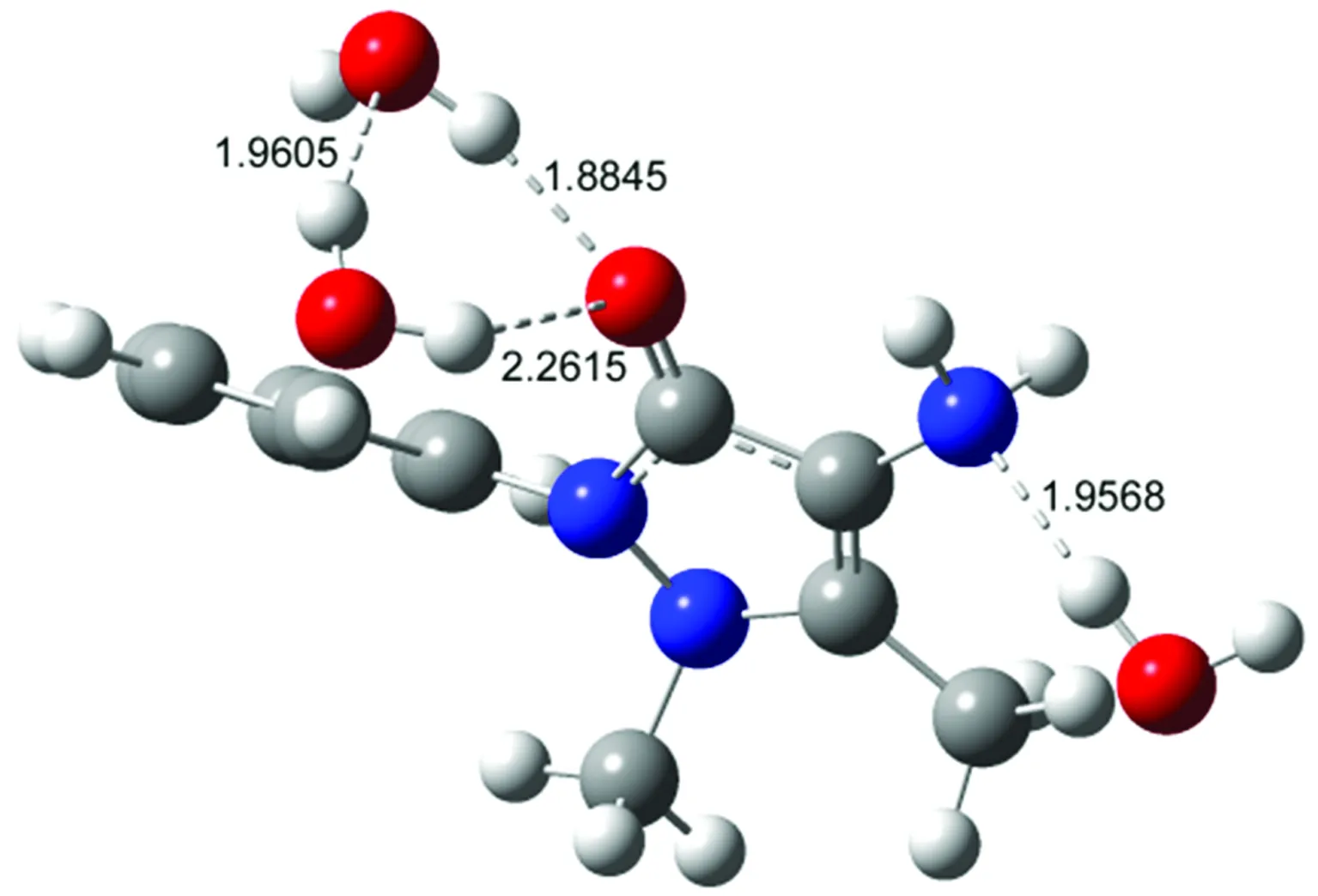
图6 水溶液中4-AAP与水分子氢键复合物结构
Fig.6Structureofhydrogenbondingcomplexof4-AAPandwatermoleculesinaqueoussolution
为进一步了解水溶液环境中溶剂分子对4-AAP电子光谱的影响,使用含时密度泛函(TD-PBE0/6-311++G(d, p))模拟了4-AAP溶剂团簇模型4-AAP-(H2O)3的电子光谱,同时使用PCM模型考虑外围溶剂的影响,得到其理论最强吸收峰波长为251 nm,在277 nm的长波长处出现一个次强吸收峰,对应的轨道跃迁贡献分别为HOMO-1→LUMO和HOMO→LUMO轨道电子跃迁。与4-AAP分子电子光谱相比,最强峰和次强峰位置均向短波长方向发生蓝移,与实验测得的吸收光谱吸收峰位置(242、274 nm)吻合,较好地解释了实验光谱中水溶液环境对于4-AAP吸收光谱的溶剂效应影响,这表明当研究4-AAP化合物在水溶液中的电子吸收光谱时,仅通过隐性溶剂PCM模型不足以准确考虑溶剂化效应的影响,其与溶剂水分子间的氢键相互作用可显著影响其电子吸收光谱特性。
3 结语

