气相色谱-负化学源-质谱法检测水中10种全氟羧酸化合物
2019-01-05王晓研沈伟健余可垚胡国绅杨功俊
王晓研, 沈伟健, 王 红, 余可垚, 吴 斌, 胡国绅, 杨功俊*
(1. 中国药科大学药学院, 江苏 南京 210009; 2. 南京海关动植物与食品检测中心, 江苏 南京 210009)
全氟化合物(PFCs)是一系列人工合成的,碳链上的氢原子全部被氟原子取代的有机化合物,因其独特的疏水疏油性被广泛用于纺织、造纸、包装等领域[1,2]。毒理学研究表明,PFCs可影响新陈代谢和生殖系统,并造成肝脏毒性[2]。目前,环境中存在的全氟化合物主要有全氟烷基羧酸类(PFCAs)、全氟烷基磺酸类(PFSAs)、全氟烷基磺酰胺类(PFOSAs)、全氟调聚醇(FTOHs)、全氟磷酸及其酯等。其中,全氟辛烷磺酸(PFOS)和全氟辛酸(PFOA)是环境中存在的最典型的两种全氟化合物,也是其他PFCs在环境中转化的最终产物。全氟化合物普遍具有难光解、水解和生物降解的特点,因此许多全氟化合物具有环境持久性,并可沿生物链积累放大[3]。联合国经济合作发展组织(OECD)及欧盟(EU)分别于2002年和2006年,将PFOS及其盐和全氟辛烷磺酰氟认定为持久性有机污染物。2009年,联合国斯德哥尔摩公约(Stockholm Convention)会议,正式将PFOS及其盐和全氟辛烷磺酰氟正式列入有机污染物名单[3]。我国也在最新的食品安全国家标准[4]中对食品接触材料及制品中PFOS和PFOA的测定做出具体规定(检出限为1.0 ng/g,定量限为2.0 ng/g)。2018年6月,美国卫生和公共服务部曁有毒物质与疾病登记处(ASTDR)公布了有关全氟烷基化合物毒性概况的草案。
目前,全氟化合物的检测方法主要包括气相色谱-电子捕获法(GC-ECD)[5]、气相色谱-质谱法(GC-MS[6-8])、气相色谱-串联质谱法(GC-MS/MS[9])和液相色谱-串联质谱法(LC-MS/MS)[10-17]。液相色谱法具有灵敏度高、检出限低、对前处理要求不高等优点,已广泛用于全氟羧酸化合物的检测。但LC系统中存在潜在的污染问题,比如PFOA是聚四氟乙烯(PTFE)合成的助剂,而在LC系统中很多管线都是PTFE材质,因此在仪器分析时容易引起本底背景值的升高,加之密封垫上含有氟聚合物涂层等,使液相色谱法难以获得理想的空白分析结果,且液相色谱的检测费用较高。气相色谱法具有检测成本低、更为普及的优势。使用气相色谱法检测全氟羧酸时[5-9],通常采用电子捕获检测器[5]和质谱检测器[6-9],两种均可达到μg/L的灵敏度。但在实际样品的检测过程中,GC-ECD检测往往只作为样品初筛的工具,阳性样品还需要质谱法进行进一步确证。相比于液相色谱法和气相色谱-电子捕获法,气相色谱-质谱法具有检测成本低,灵敏度高以及准确度高的优势。
由于全氟化合物极性大,沸点高,难挥发,因此不可直接进样,要经过衍生化的过程[18]。目前,用于全氟化合物的衍生化方法主要包括硅烷化[7]、酯化[8]、柱前衍生[5]等。如Lv等[7]利用N,O-双(三甲基硅基)三氟乙酰胺(BSTFA)衍生PFOA和PFOS,衍生化反应在40 ℃条件下反应1 h即可完成。Ingrid等[8]通过异丙醇-硫酸与PFOS发生酯化反应,从而对PFOS进行衍生,并优化了反应中硫酸的体积分数、反应时间、提取溶剂等条件。上述衍生化反应多针对单一全氟羧酸化合物,使用同一种衍生化试剂同时衍生多种全氟羧酸化合物的报道则较少。因此选择合适的衍生化试剂同时在合适的反应条件下对多种全氟羧酸化合物进行衍生,使所有化合物均达到最佳反应条件是气相色谱法用于检测全氟羧酸化合物的难点之一。
水样中含有较多杂质,需要对其进行净化以除去杂质干扰,同时对全氟羧酸化合物进行富集。本文采用弱阴离子固相萃取柱对水样进行净化富集,然后用N-甲基-N-三甲基硅基三氟乙酰胺(MSTFA)对10种全氟羧酸化合物进行衍生化,通过硅烷化降低全氟羧酸化合物的极性,并优化了反应温度、反应时间等条件,使10种全氟羧酸化合物全部出峰且峰形良好。目前,在使用气相色谱-质谱法检测全氟化合物的文献中,全部使用的是电子轰击(EI)源,考虑到全氟化合物分子结构上含有多个氟原子,电负性很强,因此负化学源(NCI)是很好的选择。本文对比了EI源和NCI源在检测全氟羧酸化合物时的优劣,结果表明在使用NCI源检测全氟化合物时,灵敏度较高且基质干扰较少。
1 实验部分
1.1 仪器、试剂与材料
7890A/5975C气相色谱-质谱联用仪(配有CI源)、7890B/5977A气相色谱-质谱联用仪(配有EI源)(美国Agilent公司); WX-80A微型涡旋混合仪(上海沪西分析仪器厂有限公司); Milli-Q去离子水发生器(美国Millipore公司); NH 03079氮吹浓缩仪(美国Horizon XcelVap公司)。
标准品:全氟丁酸(PFBA)、全氟庚酸(PFHpA)、PFOA、全氟壬酸(PFNA)、全氟癸酸(PFDA)、全氟十一酸(PFUnA)、全氟十二酸(PFDoA)、全氟十三酸(PFTA)、全氟十四酸(PFTeDA)、全氟十六酸(PFHxDA)、全氟-[1,2,3,4-13C4]-辛酸,纯度均大于90.0%,购自加拿大Wellington公司。正己烷、丙酮、乙腈、乙酸乙酯、环己烷、二氯甲烷和甲醇均为色谱纯,购自美国Burik & Jackson公司;MSTFA(含有1%(v/v)三甲基氯硅烷)购自上海源叶生物有限公司;CNW poly-sery PWAX固相萃取柱(150 mg/6 mL)购自德国CNW Technologies公司。
水样来源:南京市内河水、自来水和饮用水。
1.2 标准溶液的配制
准确称取10 mg(精确至0.1 mg)10种标准品,分别用丙酮定容至10 mL,配制成质量浓度均为1.0 g/L的标准储备液,于4 ℃冷藏保存。分别移取100 μL上述标准储备液,置于10 mL容量瓶中,用丙酮稀释并定容至刻度,配制成质量浓度为100 mg/L的混合标准工作液;用丙酮逐级稀释,配制成0.1、0.5、1.0、5.0和10.0 mg/L的系列混合标准工作液。
准确称取1.0 mg全氟-[1,2,3,4-13C4]-辛酸,用丙酮定容至10 mL,配制成质量浓度为0.1 g/L的内标储备液。移取100 μL内标储备液,置于10 mL容量瓶中,用丙酮稀释并定容至刻度,配制成10 mg/L的内标使用液。
1.3 样品前处理
取20 mL待测水样,加入100 μL 10 mg/L内标使用液,过0.45 μm聚四氟乙烯滤膜,依次用0.5%(v/v)氨水甲醇溶液、甲醇和去离子水各4 mL活化CNW poly-sery PWAX固相萃取柱(2滴/s),用稀盐酸和氢氧化钠溶液调整水样的pH值至8.0,然后用4 mL 40%(v/v)甲醇水溶液(pH 5)淋洗,抽干,最后用4 mL 0.5%(v/v)氨水甲醇溶液洗脱。
衍生:将上述4 mL洗脱液吹干,加入100 μL MSTFA(含有1%(v/v)三甲基氯硅烷),涡旋,于60 ℃水浴中超声30 min,冷却,加入900 μL乙酸乙酯,混合均匀后,供GC-NCI-MS测定。
1.4 分析条件
1.4.1GC-NCI-MS条件
色谱柱:J&W HP-5ms柱(30 m×250 μm×0.25 μm);进样口温度:280 ℃;程序升温:起始温度40 ℃,以10 ℃/min的升温速率升至115 ℃,再以2 ℃/min的升温速率升至125 ℃,最后以25 ℃/min的升温速率升至250 ℃;载气:高纯氦气(纯度≥99.999%);载气流速:0.8 mL/min;进样量:1 μL;进样方式:不分流进样。
离子源:NCI源;离子源温度:250 ℃;反应气:甲烷(纯度≥99.999%);电离能量:235 eV;辅助加热区温度:280 ℃;四极杆温度:150 ℃;溶剂延迟:3 min;扫描方式:选择离子监测模式。10种全氟羧酸化合物衍生化产物的质谱扫描参数见表1。
1.4.2GC-EI-MS条件
色谱柱:J&W HP-5ms柱(30 m×250 μm×0.25 μm);进样口温度:280 ℃;程序升温:起始温度30 ℃,保持2 min,以10 ℃/min的升温速率升至70 ℃,保持1 min,最后以15 ℃/min的升温速率升至250 ℃,保持2 min;载气:高纯氦气(纯度≥99.999%);载气流速:0.8 mL/min;进样量:1 μL;进样方式:不分流进样。
离子源:EI源;离子源温度:250 ℃;反应气:甲烷(纯度≥99.999%);电离能量:70 eV;辅助加热区温度:280 ℃;四极杆温度:150 ℃;溶剂延迟:4.3 min;扫描方式:选择离子监测模式。

表 1 10种全氟羧酸化合物和内标的保留时间和监测离子Table 1 Retention times and monitored ions of the 10 perfluorinated carboxylic acid compounds and IS
* Quantitative ion.
2 结果与讨论
2.1 质谱条件的建立
以MSTFA为衍生化试剂进行的硅烷化反应是一种重要的衍生方式,其衍生产物在EI源下的质谱碎片解析已有相关报道[19]。以全氟辛烷羧酸为例,推测其在NCI源下的质谱裂解规律见图1。在GC-NCI-MS模式下,以甲烷为反应气,在高能电子束(>70 eV)轰击下,Si-O键断裂,形成m/z为414.0和413.0的离子,这两个离子不稳定,继续脱去一个氟原子形成环氧键(m/z=394.0)或连续脱去两个氟原子形成双键(m/z=376.0)。全氟辛烷羧酸衍生化产物的质谱图如图2所示,可知推测的碎片离子与实验获得的全氟辛烷羧酸的质谱图碎片一致,因此实验选择m/z为394.0、395.0和376.0的离子作为全氟辛烷羧酸衍生化产物的质谱监测离子。其余9种全氟羧酸化合物与全氟辛烷羧酸结构相似,只是碳链长度不同,其质谱裂解机理相同,且与实验获得的质谱碎片一致。

图 1 全氟辛烷羧酸衍生化产物的质谱裂解图Fig. 1 MS fragmentation diagram of derivative of PFOA

图 2 全氟辛烷羧酸衍生化产物的质谱图Fig. 2 Mass spectrum of derivative of PFOA
2.2 衍生化反应条件的优化
2.2.1衍生化试剂的选择
全氟化合物极性较大,沸点较高,不易气化,需要进行衍生化反应使极性和沸点得到有效降低,从而适用于气相色谱分析。目前文献报道的用于全氟化合物衍生化反应的有利用异丙醇和硫酸与全氟辛烷羧酸和全氟辛烷磺酸进行酯化反应[8];利用BSTFA与全氟辛烷羧酸和全氟辛烷磺酸进行硅烷化反应[7];利用2,4-二氟苯胺为衍生化试剂以及利用N,N′-二环己基碳二亚胺为脱水剂与全氟辛烷羧酸形成酰胺化合物[20]。
硅烷化反应是一类经典的衍生化反应,利用硅烷化试剂的三甲基硅基取代目标化合物的活泼氢,从而降低目标化合物的极性,增加挥发性[20]。较为常用的硅烷化反应试剂有BSTFA、N,O-双三甲基硅基乙酰胺(BSA)和MSTFA。硅烷化反应在检测多种物质方面均有应用[14-18],崔庆新等[21]用三甲基氯硅烷衍生维生素C,从而实现气相色谱-质谱联用法测定橙汁粉中的维生素C。
本实验考察了不同衍生化试剂的衍生化效果,包括异丙醇-硫酸、五氟苄基溴、BSA、BSTFA和MSTFA。实验结果显示,异丙醇-硫酸和五氟苄基溴对10种全氟羧酸化合物标准品进行衍生化后,没有检测到衍生化后的产物,可能是因为衍生化反应不成功或衍生化产物不稳定。采用BSA、BSTFA进行衍生化,衍生化杂质相对于采用MSTFA时多,对目标产物产生较多干扰,因此最终选择MSTFA对10种全氟羧酸化合物进行衍生化。以全氟辛烷羧酸为例,MSTFA对全氟羧酸类衍生化的原理图见图3。
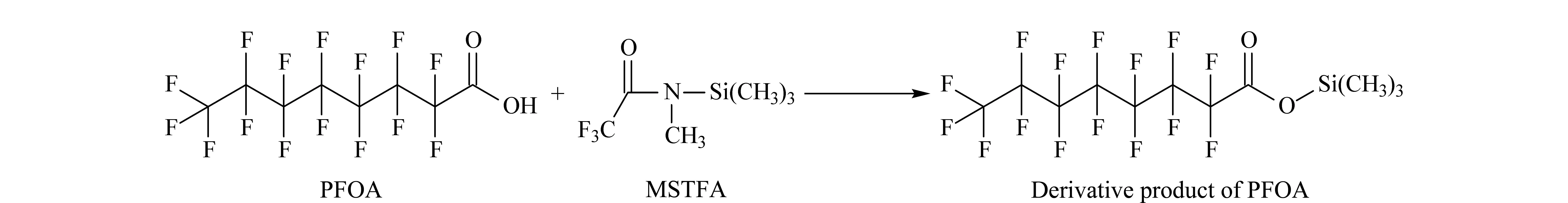
图 3 全氟辛烷羧酸的衍生化原理图Fig. 3 Derivative principle diagram of PFOAMSTFA: trifluoro-N-methyl-N-(trimethylsilyl) acetamide.
2.2.2反应时间对衍生效率的影响
取1 mL 1 mg/L的混合标准工作液,加入螺旋口玻璃小瓶中,氮气吹干后,加入100 μL MSTFA,于60 ℃反应10、20、30、40和50 min,考察不同反应时间对10种全氟羧酸化合物衍生效果的影响。结果如图4a所示,反应时间为30 min时,除PFUnA外,其余9种全氟羧酸化合物的峰面积均达到最大值。因此选取30 min作为衍生化反应的时间。

图 4 (a)反应时间、(b)反应温度、(c)超声和(d)衍生化试剂用量对衍生化产物峰面积的影响Fig. 4 Effects of (a) reaction time, (b) reaction temperature, (c) ultrasound and (d) amount of derivation agent on the peak areas of the derivative products
2.2.3反应温度对衍生效率的影响
取1 mL 1 mg/L的混合标准工作液,加入螺旋口玻璃小瓶中,氮气吹干后,加入100 μL MSTFA,分别于30、40、50, 60、70和80 ℃反应30 min,考察反应温度对衍生效果的影响。结果如图4b所示,当反应温度为60 ℃时,10种全氟羧酸化合物的峰面积均达到最大值;继续升高反应温度,峰面积反而下降。因此,60 ℃为衍生化反应的最佳温度。
2.2.4超声对衍生化效率的影响
样品经1.3节所述,洗脱液吹干后加入MSTFA,再进行简单涡旋后衍生。在此过程中,样品吹干后的残渣可能并没有与衍生化试剂充分混合,且考虑到在超声时,超声波可加快分子运动,因此考察了超声是否能提高衍生化效率,并在超声过程中用温度计监测水浴温度,将温度控制在(60±2) ℃之间。取1 mL 1 mg/L的混合标准工作液,加入螺旋口玻璃小瓶中,氮气吹干后,加入100 μL MSTFA,分别于60 ℃在超声和不超声的条件下反应30 min。结果如图4c所示,在反应过程中,超声可以明显增加衍生化反应的效率,尤其是对PFBA和PFOA而言。
2.2.5衍生化试剂用量对衍生效率的影响
取1 mL 1 mg/L的混合标准工作液,加入螺旋口玻璃小瓶中,氮气吹干后,分别加入80、100、120和140 μL的MSTFA,于60 ℃水浴超声反应30 min,比较衍生化试剂用量对衍生效率的影响。结果如图4d所示,当衍生化试剂的用量达到100 μL后,再增加衍生化试剂的用量,10种全氟羧酸的峰面积没有太大变化。因此,使用100 μL的衍生化试剂进行衍生化反应。
2.2.6溶剂对衍生化产物测定的影响
在对目标化合物进行衍生化后,衍生化产物的分子结构上增加了一个三甲基硅烷结构,有效降低了目标化合物的极性。因此,需要找到合适极性的溶剂对衍生化产物进行定容,保证10种全氟羧酸化合物的衍生化产物都可以溶解且峰形较好。实验分别比较了丙酮、乙腈、环己烷、正己烷、乙酸乙酯、甲醇和二氯甲烷7种溶剂对衍生化产物的溶解效果。结果显示,用乙酸乙酯和二氯甲烷溶解时,可检测出全部10种全氟羧酸化合物,但相对于二氯甲烷,乙酸乙酯的毒性较低。因此选择乙酸乙酯为溶剂定容衍生化产物。
2.3 前处理条件的优化
2.3.1水样的pH值
水样采用弱阴离子交换固相萃取柱CNW poly-sery PWAX进行样品前处理,以1 mg/L的混合标准溶液作为测试溶液。全氟羧酸类物质在碱性条件下为阴离子,与弱阴离子固相萃取柱实现交换,因此实验考察了水样pH值分别为7.5、8.0、8.5、9.0和9.5时,与弱阴离子固相萃取柱达到交换的程度。结果表明,当水样pH值为8.0时,10种全氟化合物的回收率均在70%~95%之间;其余条件下均出现一种或几种全氟羧酸化合物的回收率小于70%的情况。因此将水样的pH值调节至8.0。
2.3.2淋洗液甲醇的体积分数和pH值
考察了淋洗液甲醇水溶液中甲醇的体积分数及其pH值对10种全氟化合物回收率的影响。结果表明,当淋洗液pH值为5、甲醇的体积分数为40%时,10种全氟化合物的回收率均能达到70%以上,因此选择40%(v/v)甲醇水溶液(pH 5)作为淋洗液。
2.3.3洗脱液中氨水的比例
固定淋洗液为40% (v/v)甲醇水溶液(pH 5),水样pH值为8.0,考察洗脱液甲醇中氨水的体积分数(0.1%、0.5%、1.0%和1.5%)对10种全氟羧酸化合物回收率的影响。因PFHxDA的灵敏度较低,需优先考虑,当使用0.5%(v/v)氨水甲醇时,其回收率可以达到70%以上,其余9种全氟羧酸的回收率均能达到80%以上。因此,综合考虑10种全氟羧酸化合物的回收率,选择0.5%(v/v)氨水甲醇为最终的洗脱液。

图 5 采用GC-EI-MS和GC-NCI-MS时加标河水样品中 10种全氟羧酸化合物的选择离子色谱图Fig. 5 Selected ion chromatograms of the 10 perfluorinated carboxylic acid compounds in spiked river samples by GC-EI-MS and GC-NCI-MS EI: electron impact; NCI: negative chemical ionization.
2.4 离子源的选择
按照上述建立的方法对加标(10 mg/L)河水样品进行前处理,然后分别进行GC-EI-MS和GC-NCI-MS测定(见图5)。结果表明,两种方法都可以测定出10种全氟羧酸化合物,但采用GC-EI-MS时,目标峰附近出现较多杂峰,易对目标化合物的检测产生干扰;而采用GC-NCI-MS时,目标化合物周围几乎没有杂峰干扰,GC-NCI-MS的抗基质干扰能力更强。
同时比较了GC-EI-MS和GC-NCI-MS对10种全氟羧酸化合物的灵敏度。采用GC-NCI-MS时,10种全氟羧酸化合物的检出限为0.5~1.5 μg/L;而采用GC-EI-MS时,10种全氟羧酸化合物的检出限为7.5~10 μg/L,说明GC-NCI-MS对全氟羧酸类物质具有更高的灵敏度。
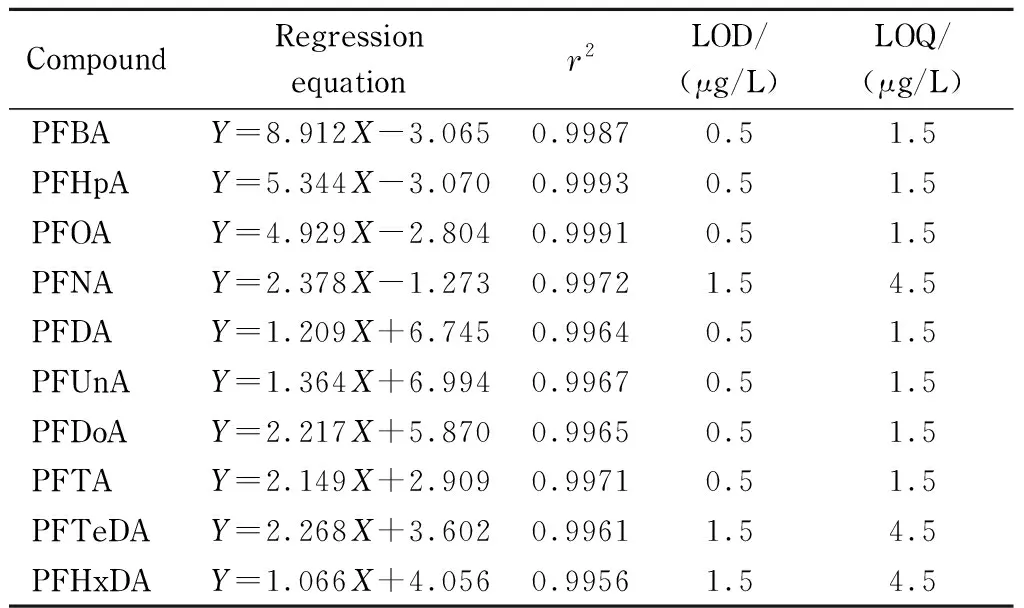
表 2 10种全氟羧酸化合物的回归方程、相关系数(r2)、 检出限和定量限Table 2 Regression equations, correlation coefficients (r2), limits of detection (LODs) and limits of quantification (LOQs) of the 10 perfluorinated carboxylic acid compounds
Y: peak area ratio of the quantitative ion of the analyte to the internal standard;X: mass concentration, mg/L.

表 3 空白加标水样中10种全氟羧酸化合物的平均 回收率和相对标准偏差(n=6)Table 3 Average recoveries and relative standard deviations (RSDs) of the 10 perfluorinated carboxylic acid compounds spiked in blank water samples (n=6)

图 6 10种全氟羧酸化合物混合标准工作液(10 mg/L) 的选择离子色谱图Fig. 6 SIM chromatogram of the 10 perfluorinated carboxylic acid compounds in a mixed standard solution (10 mg/L)
2.5 方法学评价
2.5.1线性范围、检出限和定量限
对1.2节配制的系列混合标准工作液进行分析(含内标1 mg/L), 10种全氟羧酸化合物经衍生化后的总离子流色谱图见图6。采用内标法进行定量,以各组分定量离子与内标定量离子峰面积的比值(Y)对其质量浓度(X, mg/L)进行线性回归,得到10种全氟羧酸化合物的线性方程(见表2)。10种全氟羧酸化合物的线性相关系数均大于0.995,说明10种全氟羧酸化合物在0.1~10.0 mg/L范围内线性关系良好。根据3倍和10倍信噪比分别确定10种全氟羧酸化合物的检出限和定量限,结果见表2。
2.5.2回收率与精密度
在阴性水样中分别添加100 μL质量浓度为1、10和100 mg/L的混合标准工作液(添加水平相当于0.005、0.05和0.5 mg/L),按1.3节描述进行前处理和衍生,然后在GC-NCI-MS实验条件下进样。每个添加水平平行做6个样品,每个样品重复测定两次,计算10种全氟羧酸化合物的平均回收率和相对标准偏差(RSD),结果见表3。结果表明,10种全氟羧酸化合物的平均回收率为70.2%~112.6%, RSD为2.1%~14.5%(n=6),说明建立的方法准确、可靠。
2.5.3实际样品分析
采用本文建立的方法,对南京市内河水、自来水以及饮用水样品进行检测,结果显示,在河水中检测到PFHpA、PFOA和PFDA,含量分别为2.5、4.3和6.2 ng/L;在自来水中只检测到PFOA,含量为1.3 ng/L;在饮用水中未检测到全氟羧酸类物质。
3 结论
本文采用NCI源建立了气相色谱-质谱联用测定水中10种全氟羧酸化合物的分析方法。本方法采用NCI源对全氟羧酸类化合物进行测定,结果表明,NCI源的灵敏度以及抗基质干扰能力优于EI源,线性相关性好,准确度高。为全氟羧酸化合物的检测提供了新方法。
猜你喜欢
杂志排行

色谱的其它文章
- 高效液相色谱-串联质谱法测定蔬菜水果中双甲脒及其代谢产物
- 高效液相色谱-四极杆/静电场轨道阱高分辨质谱对水产品中未知污染物的非定向快速筛查与测定
- 高效液相色谱-四极杆飞行时间质谱检测中国杨树型蜂胶、巴西绿蜂胶和杨树胶中的酚类化合物及真伪鉴别
- γ-Al2O3-氧化石墨烯吸附材料用于尿液中核苷的检测
- 基于高效液相色谱-三重四极杆质谱技术测定荔枝和香蕉中的草铵膦及3种代谢物
- Impurity profile of macitentan in Tablet dosage form using a stability-indicating high performance liquid chromatography method and forced degradation study