气相色谱法同时测定乳制品中有机氯农药和多氯联苯残留量的研究
2018-12-29李霞李洁李玲王艳丽申中兰刘艳明祝建华
李霞,李洁,李玲,王艳丽,申中兰,刘艳明,祝建华
(山东省食品药品检验研究院济南 250101)
0 引 言
乳制品中药物残留和环境污染的问题已经拉响我国奶源安全的警报。多氯联苯和有机氯农药是国际环境科学领域十分关注的持久性有机污染物。此类化合物具有亲脂性和半挥发性,难以被生物降解,可通过食物、水、大气和土壤等环境介质与包括人类在内的环境生物体系接触,对生态环境和人类的健康带来潜在的危害,因此建立多氯联苯和有机氯农药残留的检测方法对于保证乳制品的质量十分重要。
目前,多氯联苯和有机氯农药的测定对象主要有水[1-3],土壤[4],水产品[5-7],蛋及蛋制品[8],农残品[9],乳制品相对较少。多氯联苯和有机氯农药的检测方法有气相色谱法,气相色谱-质谱联用法[10]。乳制品中有机氯残留常用的净化技术有固相萃取法[11]、凝胶渗透色谱净化[12]、酸化法等,其中乳制品中有机氯残留的测定采用磺化法的报道也不多[13],乳制品中多氯联苯的测定有采用索氏提取法及自动净化处理法[14]。至今浓硫酸磺化法作为同时检测乳制品中的多氯联苯和有机氯农药残留的前处理方法尚鲜见报道。本文应用超声提取法同时提取乳制品中的多氯联苯和有机氯农药残留,采用浓硫酸净化的前处理方法,提取方法简便,浓硫酸磺化法净化效果好,回收率高,色谱背景干净,即快速又节约成本。同时也参考欧盟、日本农药残留的检测方法[15-16]和国内的标准[17-18]。所以本研究对于同时测定乳制品中有机氯和多氯联苯残留量的检测有很好的借鉴作用。
l 实验
1.1 主要仪器与试剂
气相色谱仪:Agilent 6890型,配有电子捕获检测器(FID),美国Agilent公司;旋转蒸发器:Buchi R-200型,瑞士Buchi公司;7种指示性多氯联苯混标(内含PCB28、PCB52、PCBl01、PCBll8、PCBl38、PCBl53、PCBl80,10μg/mL,纯度99.0%,溶于环己烷),德国Dr.Ehrenstorfer公司;ɑ-666、β-666、γ-666、δ-666、p,p’-DDE、o,p’-DDD、p,p’-DDD及p,p’-DDT,1 000μg/mL,纯度均大于99%,中国农业部环境保护监测所;七氯,1 000μg/mL,纯度均大于99%,中国农业部环境保护监测所;艾氏剂,1 000μg/mL,纯度均大于99%中国农业部环境保护监测所;无水硫酸钠:分析纯,于600℃下干燥4 h,冷却后储存于干燥的密闭容器中,备用;氯化钠:分析纯;乙腈:优级纯;实验所用其它试剂均为分析纯;实验用水为二次蒸馏水。
1.2 色谱条件
色谱柱:HP-1701型石英毛细管柱(30 m×0.25 mm×0.25μm);色谱柱升温程序:50℃保持1 min,20℃/min升至200℃保持1 min,5℃/min升至230℃保持5 min,5℃/min升至260℃保持10 min;进样口温度:290℃;检测器温度:300℃;载气、辅助气:均为氮气,纯度为99.999%,载气流速为3 mL/min;进样方式:不分流进样;进样量:1μL。
1.3 标准溶液的配制
分别称取适量各种多氯联苯、六六六、滴滴涕,七氯,艾氏剂,用正己烷配制成0.5μg/mL储备液。根据需要,用正己烷再配制成所需浓度的混合标准溶液。
1.4 试验方法
液体样品称取2.0 g样品于50 mL离心管中,加入2 g氯化钠,加入20 mL乙腈提取液,涡旋2 min,超声30 min,再加入无水硫酸钠5 g脱水,涡旋2 min,于8 000 r/min离心3 min,将上清液转移到50 mL旋转蒸发瓶中,下层样品再提取一次后合并上清液,40℃水浴中旋转蒸至近干,加入10 mL正己烷复溶,再加入1 mL硫酸磺化,于8 000 r/min离心3 min,后取上清液氮气吹干,准确加入1 mL正己烷复溶,再加入0.2 mL浓硫酸磺化,8 000 r/min离心3 min,取上清液用纯净水洗至中性,取上清液进入气相色谱分析。
固体样品称取2.0 g样品于50 mL离心管中,加入10 mL水,涡旋0.5 min,加入2 g氯化钠,再加入20 mL乙腈提取液,涡旋2 min,超声30 min,放入离心机,8 000 r/min离心3 min,取上清液于50 mL离心管中,加入5 g无水硫酸钠脱水,转移到50 mL旋转蒸发瓶中,下层样品再提取一次后合并上清液,40℃水浴中旋转蒸至近干,加入10 mL正己烷复溶,加入1 mL硫酸磺化,于8 000 r/min离心3 min,后取上清液氮气吹干,准确加入1 mL正己烷复溶,再加入0.2 mL浓硫酸磺化,8 000 r/min离心3 min,取上清液用纯净水洗至中性,取上清液进入气相色谱分析。
2 结果与讨论
2.1 提取条件的优化
2.1.1 提取溶剂的选择
六六六、滴滴涕、七氯、狄试剂和多氯联苯属于极性物质,乳制品中脂类物质是非极性物质,根据相似相容原理,选用极性有机溶剂提取目标物可以减少脂类物质的干扰。实验对乙腈、甲醇两种极性提取溶剂进行考察。结果表明:乙腈作为提取溶剂时,乳制品中4种六六六、4种滴滴涕、七氯、狄试剂和7种多氯联苯的加标回收率比甲醇作为提取溶剂时得到的加标回收率好,所以选用乙腈为本实验的最佳提取溶剂。
2.1.2 提取方法
提取方法的选择实验以鲜牛奶样品的加标回收率来考察超声提取法,分别以超声10 min,20 min,30 min,40 min进行考察。17种化合物的加标回收率如图1所示。超声10 min的加标回收率在70.58%~81.68%,超声20 min的加标回收率在75.63%~88.23%,超声30 min的加标回收率在83.55%~95.38%,超声40 min加标回收率在83.6%~95.57%,超声30 min和超声40 min两者相差不大,考虑到试验时间的节约,所以选择超声30 min提取法的最佳超声时间。
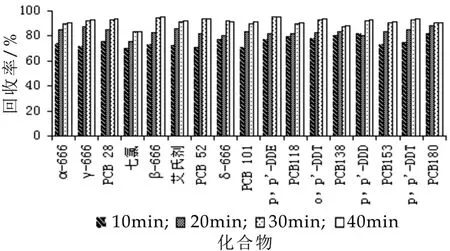
图1 不同超声时间的回收率
2.1.3 净化条件的优化
以鲜牛奶样品为对象来考察浓硫酸用量,分别加入量为0.5、1.0、2.0 mL的浓硫酸进行考察,不同用量浓硫酸对空白鲜牛奶样品的净化效果见图2。实验结果表明:在加入0.5 mL浓硫酸后,经涡旋、离心,提取物溶液发生分层但上层溶液仍存在一些黄色的脂类物质,而当加入1.0 mL和2.0 mL的浓硫酸时,提取物溶液发生分层明显且上清液澄清,有较好的净化作用,同时考虑溶剂的节约,所以选择浓硫酸的用量为1.0 mL。加标量为10μg/kg的鲜牛奶、酸牛奶和奶粉的净化效果如图3。在实验过程中,单独使用浓硫酸净化后会产生离子型杂质及浓硫酸的残留,这些物质会污染气相色谱仪的毛细管柱,且目标物有干扰峰出现,而加蒸馏水洗至中性后对气相色谱仪的毛细管柱起到保护作用,并且化合物的回收率不受影响。加蒸馏水水洗和不加水洗的效果图如图4。
2.1.4 色谱柱的选择
本实验在确定进样口温度、检测器温度、载气流速及其他相关条件一致的情况下,实验中采用HP-5(30 m×0.25 mm×0.25μm)柱子无法使其中的某些化合物分开。经过多次试验,试验中采用DB-1701(30 m×0.25 mm×0.25μm)的色谱柱可以使所有化合物达到很好的分离,并且在保证分离效果的前提下,改变升温程序还可以缩短分析样品所需时间。所得色谱最佳分离条件如前所述,色谱分离效果见图2。

图2 不同浓硫酸加入量对空白鲜牛奶样品的净化效果
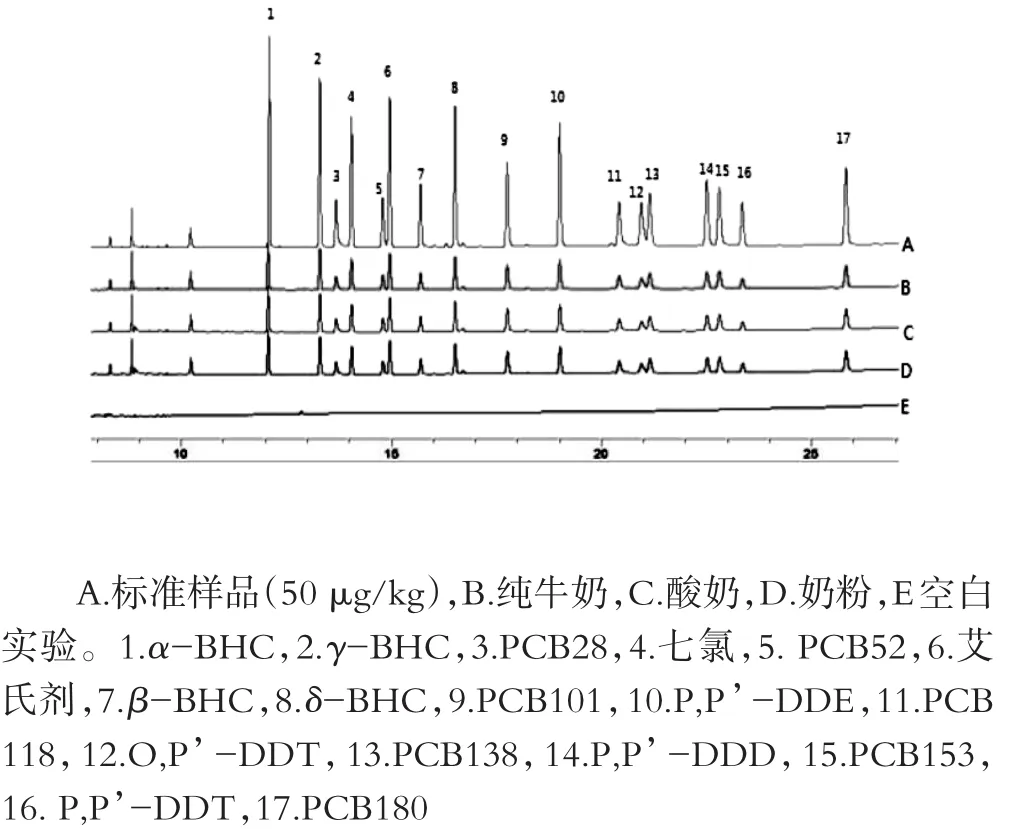
图3 加标量(10μg/kg)鲜牛奶、酸牛奶及奶粉的的净化效果

图4 加标量(10μg/kg)鲜牛奶加蒸馏水水洗及不加蒸馏水水洗的效果
2.2 方法的考察
2.2.1 线性范围、检出限和定量限
实验以空白鲜牛奶样品为基质,各称取鲜牛奶2.0 g于一系列50 mL具塞离心管中,1.4节的方法进行前处理得到空白样品基质,依次加入待测组分得到质量浓度为5、10、20、40、100、200μg/L的一系列基质标准溶液,按1.2节的分析方法测定,采用外标法定量。以峰面积为纵坐标(y),目标物的质量浓度为横坐标(x,μg/L)绘制基质标准曲线,线性范围结果见表1。方法的检出限和定量限范围分别为0.08~0.27μg/kg和0.27~0.90μg/kg,可满足乳制品中4种六六六、4种滴滴涕、七氯、艾氏剂和7种指示性多氯联苯残留分析的要求。实验中以所得到的3倍信噪比(RSN=3)计算检出限,以所得到的10倍信噪比(RSN=10)计算定量限,检出限和定量限的结果见表1。
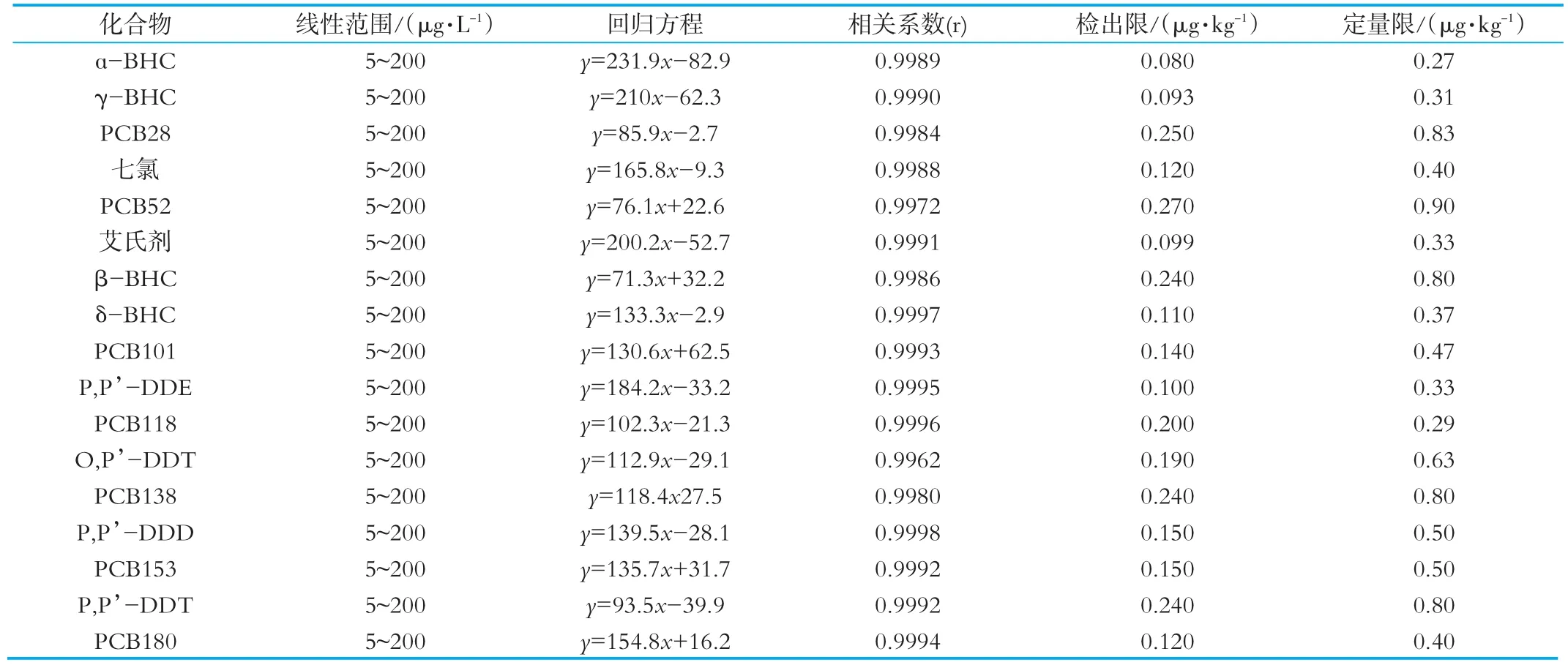
表1 4种六六六、4种滴滴涕、七氯、艾氏剂和7种指示性多氯联苯的线性范围、线性方程、相关系数(r)、检出限
2.2.2 加标回收率
实验分别加标量为5、10、50 mg/kg的鲜牛奶、奶粉和酸奶(每个水平重复6次),按照1.2节和1.4节的方法进行前处理和分析测定,计算加标回收率和RSD,结果见表2-4表。

表2 纯牛奶中添加4种六六六、4种滴滴涕、七氯、艾氏剂和7种多氧联苯的加标回收率和RSD(n=6)

表3 酸奶中添加4种六六六、4种滴滴涕、七氯、艾氏剂和7种多氧联苯的加标回收率和RSD(n=6)

表4 奶粉中添加4种六六六、4种滴滴涕、七氯、艾氏剂和7种性多氧联苯的加标回收率和RSD(n=6)
表2-表4表明:在10、20、50 mg/kg 3个加标水平下,纯牛奶空白样品中17种化合物的加标回收率范围为87.13%~99.40%,RSD范围为2.17%~7.67%;酸奶空白样品中17种化合物的加标回收率范围为85.09%~97.65%,RSD范围为1.23%~8.12%;奶粉空白样品中17种化合物的加标回收率范围为86.40%~97.93%,RSD范围为1.89%~7.04%。该方法的准确度与精密度高,符合鲜牛奶、酸奶和奶粉中4种六六六、4种滴滴涕、七氯、艾氏剂和7种指示性多氯联苯残留分析的要求。
2.2.3 定量方法的选择
实验表明:使用溶剂标准曲线对乳制品样品中4种六六六、4种滴滴涕、七氯、艾氏剂和7种多氯联苯进行定量所得到的加标回收率均超出110%,这是因为在气相色谱分析中这些化合物表现出的不同程度的基质增强效应,使得待测物检测信号增强。为了提高方法的可靠性和结果的准确度,消除基质效应,在实验室中采用不含待测物的空白样品经过1.4节方法进行处理,加入与溶剂标准曲线相同系列浓度的待测组分,即制作基质标准曲线来进行定量分析。实验中分别对纯牛奶,酸牛奶和奶粉配制了基质标准曲线,结果表明,三种样品空白基质曲线中的待测组分的响应值和标准曲线的斜率基本相同,所以为了避免每个样品种类都要配制其基质匹配标准曲线,实验中选取相对杂质较少的鲜牛奶空白基质作基质曲线。实验表明使用基质标准曲线进行定量得到的加标回收率在80%~110%之间,消除了基质效应的影响,同时提高方法的可靠性和结果的准确度。
2.2.4 实际样品的检测
实验中对随机送检的30批乳制品分别按照1.2节和1.4节的方法进行前处理和分析测定,结果表明:纯牛奶,酸奶和奶粉中的4种六六六、4种滴滴涕、七氯,艾氏剂和7种指示性多氯联苯均未检出。
3 结论
本实验建立了浓硫酸净化的前处理方法,在乳制品六六六、滴滴涕,七氯,艾氏剂和指示性多氯联苯的检测中取得较好的净化效果,并且结合气相色谱法实现对乳制品中六六六、滴滴涕,七氯,艾氏剂和指示性多氯联苯的同时定量分析,适用于乳制品中六六六、滴滴涕,七氯,艾氏剂和指示性多氯联苯的快速检测。
