伴有视网膜病-肾病-卒中的遗传性内皮细胞病一例
2018-12-21俞英欣姚生戚晓昆钱海蓉李媛
俞英欣 姚生 戚晓昆 钱海蓉 李媛
视网膜病-肾病-卒中的遗传性内皮细胞病(hereditary endotheliopathy with retinopathy,nephropathy and stroke,HERNS)是一种非常少见的遗传性脑小血管病。脑小血管疾病是指颅内小动脉和微动脉病变导致脑卒中发作的一组疾病。通常累及直径<300 μm的动脉血管。遗传性脑小血管病是指有明确遗传学机制的家族性脑小血管病,如伴皮质下梗死和白质脑病的常染色体隐性遗传性脑动脉病(CARASIL)、伴皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病(CADASIL)、弥漫性躯体血管角质瘤(Fabry病)、视网膜血管病变伴有大脑白质脑病(retinal vasculopathy with cerebralleukodystrophy,RVCL)等。其中RVCL包括伴有HERNS、遗传性血管性视网膜病(hereditary vascular retinal disease, HVR)、大脑视网膜血管病(cerebral retinavascular disease ,CRV)和遗传性系统性脑血管病(hereditary systemic angiopathy ,HSA)。迄今HERNS病国内外均少见报道,由Jen等[1]在1997年首次报道了一个家系,我国于2007年张在强等[2]报道1例。本文对作者医院经活检证实的1例HERNS进行报道。
1病例报道患者男性,39岁,已婚,因“视物不清,尿蛋白及左侧肢体力弱5年,言语减少伴右侧肢体无力1个月”于2017-01-09入作者医院。患者2012年自觉休息欠佳时出现头脑不清晰,无头晕,头痛,反应迟钝等症状,未在意。2012-04自觉右眼视物发暗及模糊,当地医院眼科完善检查诊断为“视神经损伤、高血压、尿蛋白”,遂就诊于肾内科,当时头颅MRI检查提示右额叶T1低信号,T2高信号,病灶呈环形强化(图1A-B)。对症治疗后于2012-07-16复查头颅MRI提示右额叶病灶增大并有强化(图1C-D)。遂于2012-07-19在外院行右额病变切除术,术后病理结果示:(右额叶)送检破碎脑组织块,镜下可见神经元分布区枝丫状血管增生伴轻度淤血,部分神经元变性,白质疏松,胶质细胞及小血管增生,部分血管壁增厚玻变,周围少量淋巴细胞浸润,并伴有白质内多灶性变性坏死,少量格子细胞形成,局灶蛛网膜下腔还可见一小团畸形血管丛,符合小血管病变导致局灶脑梗死改变。后患者按脑梗死给予抗血小板聚集,强化他汀,改善循环等治疗,症状无明显加重。患者于2013—2015年间曾多次复查头颅MRI及尿常规,尿蛋白持续(++)。于2015-02发现血肌酐增加,于外院行肾活检病理(2015-3-22)提示:膜增殖性肾小球肾炎;缺血性肾病,遂服用醋酸泼尼松片40 mg,并逐渐减量;2015-06复查肾功能较前有所好转;于2015-06上呼吸道感染后出现“肺部真菌感染”,于当地医院住院治疗好转;2015-10-01出现便血,外院考虑“消化道出血”,治疗2周无明显好转,出血量增大,考虑消化道大出血,行手术治疗,术中发现肠道多发溃疡,诊断为“小肠溃疡”,切除部分小肠以及对症治疗后病情平稳。2016-11患者逐渐出现淡漠,言语少,找词困难,行动迟缓, 2016-12-16症状明显加重,几乎不能言语,能理解家人说话,并出现右侧肢体无力及肿胀,以右上肢为著,外院行头颅MRI检查示:右额叶术后改变,左额颞占位性病变,长T1长T2信号,占位效应明显,无明显强化,中线移位,脑室受压(图2A-D),为求进一步诊治,于2016-12-20入作者医院神经外科,2016-12-27全麻下行“导航下左额病变切除术”, 脑组织病理(2017-01-03)示:脑组织大片坏死形成,中性粒细胞及组织细胞反应,小血管闭塞,血管壁纤维素变性坏死,周围胶质细胞增生(图3A-D),以上病变符合脑梗死。术后按脑梗死治疗,转入作者医院神经内科。既往史:否认高血压,糖尿病,高脂血症等病史。父母体健,否认家族遗传病病史。
神经系统查体:意识清楚,运动性失语,强哭强笑,右眼对色彩感应差,双侧掌颏反射阳性,四肢肌张力稍高,左侧肢体肌力Ⅴ-级,右侧上肢肌力Ⅰ级,右侧下肢肌Ⅲ级,四肢腱反射活跃,双侧Babinski征、Chaddock征阳性,感觉查体不能配合。颈软,无抵抗。
实验室检查示:补体C3728 mg/L,红细胞2.42×1012/L,血红蛋白72 g/L,红细胞压积0.225 g/L,白细胞计数11.54×109/L,血清白蛋白30.0 g/L,血尿素11.0 mmol/L,血尿酸632.0 μmol/L,血肌酐215.4 μmol/L,血清铁9.5 μmol/L,叶酸(发光法)5.22 nmol/L,维生素B1299 pmol/L,D-D二聚体1317.0 ng/mL,同型半胱氨酸40.4 μmol/L,总三碘甲状腺原氨酸(TT3)0.85 nmol/L、促甲状腺激素(TSH)5.17 mIU/L,尿蛋白 0.25 g/L(+)。肺CT(2017-01-13)示:双下肺透过度不均并散在索条、条片影,考虑慢性炎症可能性大;右肺中叶小钙化结节灶;双侧胸膜稍增厚。头颅MRA未见明显异常(图4A),眼底照相未见明显异常(图4C-D),因患者肾功能异常,未能行眼底荧光造影。术后2017-02-06复查头颅CT,提示双额叶低密度病灶(图4 B )。

图1 患者头颅MRI表现:2012-06-26右侧额叶可见长T2信号病灶(A),呈环形强化(B);2012-07-16头颅MRI右侧额叶病灶呈环形强化(C),右额叶病灶增大并有强化(D)

图2 患者左额脑病变切除术后头颅MRI(2017-1-12): DWI示左额脑病变切除术后改变,左额叶DWI高信号病灶(A), FLAIR示左侧额叶术区轻度脑膜脑组织膨出,硬膜外少量积液、血肿;左额叶术区周围混杂信号(B); T2示左侧额叶术区轻度脑膜脑组织膨出,硬膜外少量积液、血肿;左额叶术区周围混杂信号(C);强化示左额叶病灶环形强化(D)
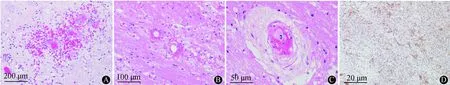
图3 患者左额叶脑组织病理示:脑组织HE染色低倍镜下示血管壁纤维素样坏死,继发出血(A);较高倍镜下示血管周围炎性细胞浸润,反应性星形胶质细胞增生(B),高倍镜下示动脉小血管坏死(C); GFAP染色提示胶质细胞增生(D)

图4 患者左额病变切除术前、术后影像学表现:术前头颅MRA未见明显异常(A), 术后头颅CT示左侧额叶软化灶,术后改变,右侧额叶低密度灶,肿胀明显,左额叶去骨瓣(B);术前眼底照相均未见明显异常(C图为左侧,D图为右侧)
该患者及其父母进行了包括NOTCH3、HTRA1、COL4A1、KRIT1、PRNP、3原初核酸外切酶1(three-prime repair exonuclease-1,TREX1)基因在内的多个脑小血管病遗传基因测序。患者TREX1基因外显子区域发现两处杂合突变点:c.531C>T、c.734dupC (胞嘧啶>胸腺嘧啶,插入突变),导致氨基酸改变p.Y177Y、p.S246IfsX16(氨基酸未发生改变,丝氨酸>异亮氨酸,移码16位后终止);突变位点c.531C>T其母为纯合子突变,该位点东亚人群携带率较高,不致病;c.734dupC位点为致病性突,其父母均无该位点突变。PRNP基因测序,发现该基因外显子一处杂合突变, c.38G>A(鸟嘌呤>腺嘌呤),导致氨基酸p.V180I(缬氨酸>异亮氨酸),其父有相同突变。其他基因该患者及其父母均无突变。
结合患者的临床,影像学,病理及基因检测结果,最后诊断为HERNS病。给予对症治疗及醋酸泼尼松龙30 mg/d维持治疗。住院1月后出院,停用激素,继续抗癫痫药物及维生素治疗,2018年8月随访,病情稳定无复发。
2讨论本例病例为青年男性,主要表现为视网膜病变、高血压、缺血性肾病、反复发生的脑血管病及小肠溃疡等。实验室检查提示高尿酸血症、贫血、半胱氨酸血症、尿蛋白和慢性肺炎。诊断时需综合考虑各系统损害,详细的病史,体格检查和所有的实验室检查情况。首次头颅影像学提示突发的右侧额叶混杂性强化病灶,占位效应明显,后病灶加大,患者当时为34岁。38岁时,头颅影像学示为左侧额顶叶混杂强化病灶,占位效应明显。就诊于多家医院,均按不同系统疾病进行诊治。肾活检提示为缺血性肾病,脑组织病理学也提示缺血性脑血管病,诊断需考虑血管壁病变可能。鉴别诊断考虑自身免疫性疾病,但患者无免疫学相关指标的异常。综合患者各个脏器的损害,并检索相关文献,诊断为RVCL,HERNS。
RVCL是成人发病的常染色体显性遗传性疾病,发病年龄30~50岁,它包括CRV、HVRI、HERNS和HSA。(1)HERNS主要表现视野缺损,反复卒中发作,肾功能不全和蛋白尿以及不同程度的精神症状。1997年Jen等[1]报道了一个HERNS病华裔家系,该家系3代11人发病,表现为中枢神经系统和视网膜血管病,常染色体显性遗传。Jen等将其命名为遗传性内皮细胞病伴视网膜病、肾病和卒中(HERNS)。2007年张在强等[2]报道一例HERNS病病例。该患者早期表现为视觉损害和肾功能障碍,神经系统症状一般出现在30~50岁,呈偏头痛样头痛、并逐渐出现局灶性神经功能损害(如偏瘫、构音障碍、失用等);荧光血管造影显示视网膜血管的病变;眼科检查发现黄斑水肿伴有毛细血管排空和中央凹周毛细血管扩张。HERNS病影像学显示特征性额顶叶“假瘤征象”,皮质下白质T1低信号,T2高信号,有明显强化和病灶周围水肿[3],病理学特征性改变为脑内及其他器官(如肾、胃、大网膜、小肠和皮肤)的血管基底膜增厚,多层化改变。(2) HVR早期表现为集中在黄斑周围的视网膜微血管病变,微动脉瘤和毛细血管扩张;晚期视网膜动脉分支闭塞。80%患者存在雷诺现象,70%患者具有偏头痛,55%患者雷诺现象和偏头痛二者同时存在[4]。一般认为HVR并不影响患者预期寿命。(3)CRV患者临床表现为视网膜毛细血管变性和脑血管病变,神经系统表现为偏头痛、卒中和痴呆;脑MRI显示“假瘤征象”,并且脑内假瘤发生在50岁之前,为其主要致死原因。CRV与HERNS患者多于55岁内死于神经系统并发症[3]。
RVCL是TREX1基因突变所致,该基因位于3号染色体短臂21.3-21.2区域,全长2126 bp,包含1个外显子,编码314个氨基酸的3原初核酸外切酶1。目前报道TREX1 基因突变有37种,包括错义/无义为18种,小的缺失5种,小的插入13种,大的插入1种。TREX1突变还可以造成其他疾病,如 A ic a rd i-Goutieres 综合征1型:该病是一种遗传异质性常染色体隐性遗传性脑病。表现为脑萎缩、脑白质营养不良、颅内钙化、慢性脑脊髓液淋巴细胞增多及脑脊液α-干扰素增高等[5]。系统性红斑狼疮易感性:TREX1基因突变可造成系统性红斑狼疮易感。此类患者同时可表现为小的脑白质缺血性损害和局灶性脑梗死[6]。家族性冻疮狼疮:TREX1基因突变还可导致家族性冻疮红斑狼疮[7]。迄今为止仅有12个家族和4个独立病例的RVCL基因突变的报道,不同位点的突变有不同的临床表现[8]。TREX1突变造成RVCL的原因尚不清楚,有报道干扰素(IFN)诱导基因Ⅰ型的RVCL病例[9]。此患者母亲携带相同的基因,但并未发病,其他家族史不详亦无法追查基因突变情况。与目前报道的所有RVCL病例基因突变进行比对,此次报道的病例为新TREX1基因突变造成的HERNS病。目前该患者病史至今5年,仍在随访中。
目前对于HERNS尚无有效的治疗方法,治疗原则主要是减轻疼痛及缓解症状,可以给予阿司匹林,对于某些患者可给予激光治疗以预防肾脏出血,小剂量泼尼松维持治疗可以减轻脑水肿。RVCL的预后较差,生存期5~10年。
综上,HERNS是一种非常少见的遗传性脑小血管病,预后较差、生存期较长,尚无有效的治疗方法。临床应对该病予以关注,以防误诊。
志谢
作者衷心感谢北京301病理科桂秋萍教授在诊断中给予的大力帮助。
