蒙药材制牛鞭的质量标准研究
2018-12-19邢界红刘庆玲岳景茁邓伟明
邢界红 刘庆玲 岳景茁 邓伟明
(内蒙古自治区蒙药股份有限公司,内蒙古 通辽 028000)
1 仪器与试药
1.1 仪器 高效液相色谱仪(赛默飞,型号:U3000)、效液相色谱仪二极管阵列检测器、电子天平(十万分之一 型号:CP225D);
1.2 试剂与试药 异硫氰酸苯酯(PITC)购自北京百灵威科技有限公司 标示纯度≥98%;甘氨酸(批号 140689-201605)、丙氨酸(批号 040680-201303)、脯氨酸(批号 140677-201206)由中国食品药品生物制品鉴定所提供;乙腈、乙酸钠、盐酸、三乙胺均为分析纯,市售。
2 方法与结果
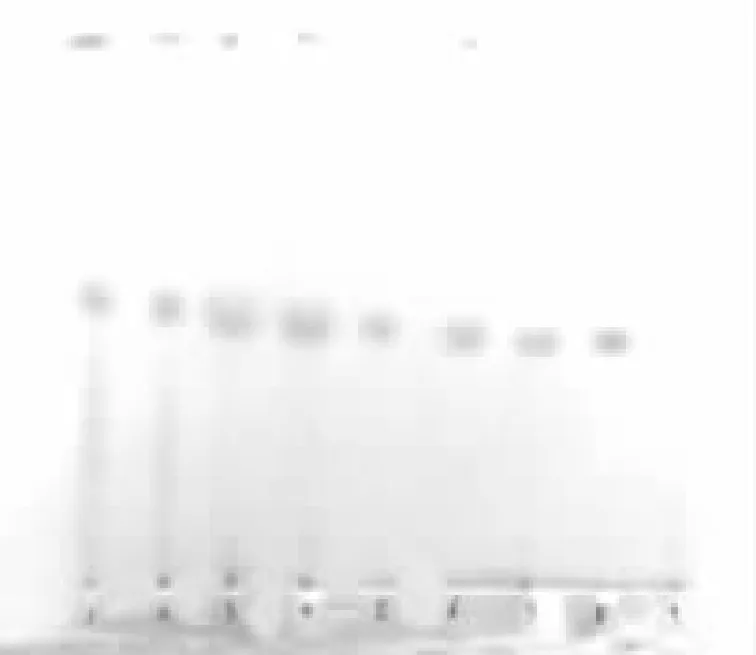
2.1 薄层鉴别 取本品粉末5g,置索氏提取器中,加乙醚70mL,加热回流30min,溶液蒸干,残渣加乙醚2mL使溶解,作为供试品溶液。另取胆固醇对照品,加乙醚制成每1mL含1mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502 )试验[1],吸取上述2种溶液各5μL,分别点于同一硅胶G薄层板上,以石油醚-丙酮(4:1)为展开剂,展开,取出,晾干,喷以乙醇-浓硫酸-醋酐(10:1:1)的溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。结果见图(5为对照品,9为阴性对照,其它为7批样品)。
2.2 浸出物 取制牛鞭(批号:201608001),分别照水溶性浸出物测定法和醇溶性浸出物测定法[2](《中国药典》2015年版四部通则2202)项下的冷浸法和热浸法测定,浸出物结果显示水热浸法43.18%,水冷浸法35.98%;95%乙醇热浸法15.60%,95%乙醇冷浸法12.30%;75%乙醇热浸法21.28%,75%乙醇冷浸法17.99%,因水溶性热浸出物含量较高且溶剂为水,故本品浸出物选择采用水溶性浸出物测定方法的热浸法测定,浸出物结果显示,7批样品水溶性热浸出物的平均值为33.03% ,最高38.99% ,最低26.01%,因此将浸出物含量暂定为不得少于25.0%。
2.3 含量测定
2.3.1 方法 照高效液相色谱法(通则0512)测定:含量=CR× Ax /ARCR为对照品浓度、Ax为供试品峰面积、 AR为对照品浓度。
2.3.2 色谱条件[3]赛默飞高效液相色谱仪 型号(μLtimate3000)紫外∕二级管阵列检测器 Chromeleon7工作站 色谱柱:①ΜLtimate plus SN:P18151625 C18(4.6mm×250mm);②Thermo SN:10306598 C18(4.6mm×250mm)③Inertsustain SN:6LR98043 C18(4.6mm×250mm);柱温43℃;以乙腈-0.lmol·L-1乙酸钠溶液(用乙酸调节p H 值至6.5)(7 : 93)为流动相A ,以乙腈-水(4 : 1)为流动相B ,按下表中的规定进行梯度洗脱;检测波长为254nm,进样量5μL。

表1 流动相
表2显示:研究组患者的残余尿量(98.6±38.5)mL较对照组(136.3±52.2)mL明显减少,切(P< 0.05),差异有统计学意义。
2.3.3 对照品溶液的制备 取甘氨酸对照品、丙氨酸对照品、脯氨酸对照品适量,精密称定,加0.lmol·L-1盐酸溶液制成每1mL含甘氨酸0.1mg·mL-1、丙氨酸对照品0.1mg·mL-1、脯氨酸0.1mg·mL-1的混合溶液,即得。
2.3.4 供试品溶液的制备 取本品(过三号筛)约0.25g,精密称定,置25mL量瓶中,加0.1mol/L盐酸溶液20mL,超声处理(功率300W,频率40kHz)30min,放冷,加0.lmol·L-1盐酸溶液至刻度,摇匀。精密量取5mL,置20 mL安瓿中,加盐酸5mL,150°C水解1h,放冷,转移至蒸发皿中,用水10mL分次洗涤,洗液并入蒸发皿中,蒸干,残渣加0.1mol·L-1盐酸溶液使溶解,转移至25mL量瓶中,加0.1mol·L-1盐酸溶液至刻度,摇匀,即得。
精密量取上述对照品溶液和供试品溶液各5mL,分别置25mL量瓶中,各加0.1mol·L-1异硫氰酸苯酯(PITC)的乙腈溶液2.5mL,lmol·L-1三乙胺的乙腈溶液2.5mL,摇匀,室温放置1 h后,加50%乙腈至刻度,摇匀。精密吸取10mL,加正己烷10mL,振摇,放置10min,取下层溶液,滤过,取续滤液,即得。
空白溶液 取0.lmol·L-1盐酸溶液5mL,精密量取上述对照品溶液和供试品溶液各5mL, “置25mL量瓶中,各加0.lmol·L-1异硫氰酸苯酯(PITC)的乙腈溶液2.5mL”起,依法测定含量,即得。
2.3.5 系统适用性试验 峰纯度试验:采用高效液相色谱仪二极管阵列检测器检测,分别精密吸取对照品溶液与供试品溶液各5μL,注入液相色谱仪,测得结果对照品与供试品色谱峰匹配值均大于95%。
分别吸取上述对照品溶液、供试品溶液及空白溶液各5μL,按色谱条件进行测定,结果样品色谱中甘氨酸、丙氨酸、L-脯氨酸3个氨基酸色谱峰分离度良好,空白无干扰。
2.3.6 提取效率的考察 超声时间考察 以0.1mol·L-1盐酸溶液作为提取溶剂进行超声外理(功率300W,频率40kHz),为保证被测成分提取完全,实验中考察了超声提取20min、30min、40min不同时间对提取效率的影呴,结果表明超声处理20min、30min和40min含量基本一致,故超声时间定为30min。
酸水解时间考察 在提取过程中,对柱前酸水解时间进行考察,结果可见,超声水解30min、60min和90min含量基本一致,故水解时间定为60min。
2.3.7 线性关系考察 分别吸取上述对照品溶液2μL、5μL、8μL、10μL、12μL、15μL注入液相色谱仪,测得峰面积。分别以氨基酸对照品的进样量为横坐标,峰面积为纵坐标,绘制标准曲线,计算回归方程。结果见下表2~4。
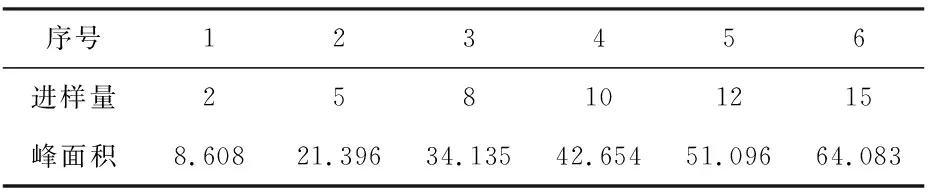
表2 线性关系考察—甘氨酸

表3 线性关系考察—丙氨酸
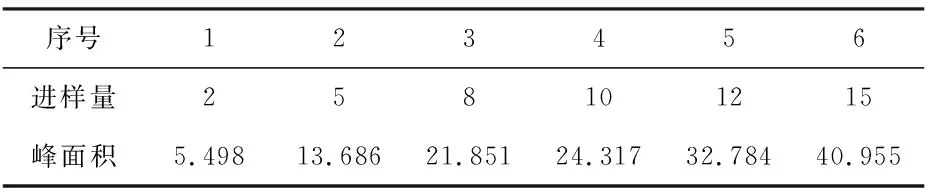
表4 线性关系考察—脯氨酸
分别以氨基酸对照品的进样量为横坐标,峰面积为纵坐标,绘制标准曲线,计算回归方程。从而得出二者具有极好的线性相关性,相关系数r均大于0.999。
2.3.8 稳定性 取供试品按上述色谱条件分别于0.3.6.9.12.h测定峰面积计算甘氨酸、丙氨酸、脯氨酸3个氨基酸色谱峰峰面积的RSD分别为0.08%、0.32%、1.77% ,供试品溶液在12h内稳定。
2.3.9 精密度 取供试品溶液按照上述色谱条件连续进样6针,测定峰面积甘氨酸、丙氨酸、脯氨酸3个氨基酸的RSD分别为0.05%、0.09%、0.68%。结果表明,仪器的精密度良好。
2.3.10 重复性 取同一批次的制牛鞭6份,按含量测定项下方法操作,测定每份样品的含量,甘氨酸平均含量40.73 mg·g-1, RSD%=0.56%;丙氨酸平均含量22.09 mg·g-1, RSD%=1.69%;脯氨酸平均含量23.54 mg·g-1, RSD%=0.37。
2.3.11 加样回收试验 取甘氨酸对照品加0.1mol·L-1盐酸制成每1mL含2.396mg的溶液,取丙氨酸对照品加0.1mol·L-1盐酸制成每1mL含1.397mg的溶液,取甘氨酸对照品加0.1mol·L-1盐酸制成每1mL含1.702mg的溶液,另取已知含量制牛鞭样品(批号:201608005,甘氨酸含量40.73mg·g-1、丙氨酸含量22.09 mg·g-1、脯氨酸含量23.54 mg·g-1)约0.125g,共6份,精密称定,分别加入上述甘氨酸对照品溶液2mL、丙氨酸对照品溶液3mL、脯氨酸对照品溶液2mL(约相当于供试品含量的100%),照前述方法测定,并计算回收率,结果见表5~7。

表5 加样回收试验结果-甘氨酸

表6 加样回收试验结果-丙氨酸

表7 加样回收试验结果-脯氨酸
2.3.12 样品测定 取制牛鞭样品0.25g,精密称定,按上述方法操作,并按干燥品计算含量。制牛鞭7批样品的含量测定结果见表8~10。
样品测定结果表明,7批样品甘氨酸平均含量为38.98mg·g-1,丙氨酸平均含量26.75mg·g-1,脯氨酸平均含量22.65mg·g-1,按平均值的80%(-20%)制定含量限度甘氨酸31.18mg·g-1,丙氨酸21.4 mg·g-1,脯氨酸18.12 mg·g-1,取整数,限度暂定为甘氨酸30.0 mg·g-1,丙氨酸20.0 mg·g-1,脯氨酸18.0 mg·g-1。

表8 样品测定试验—甘氨酸

表9 样品测定试验—丙氨酸
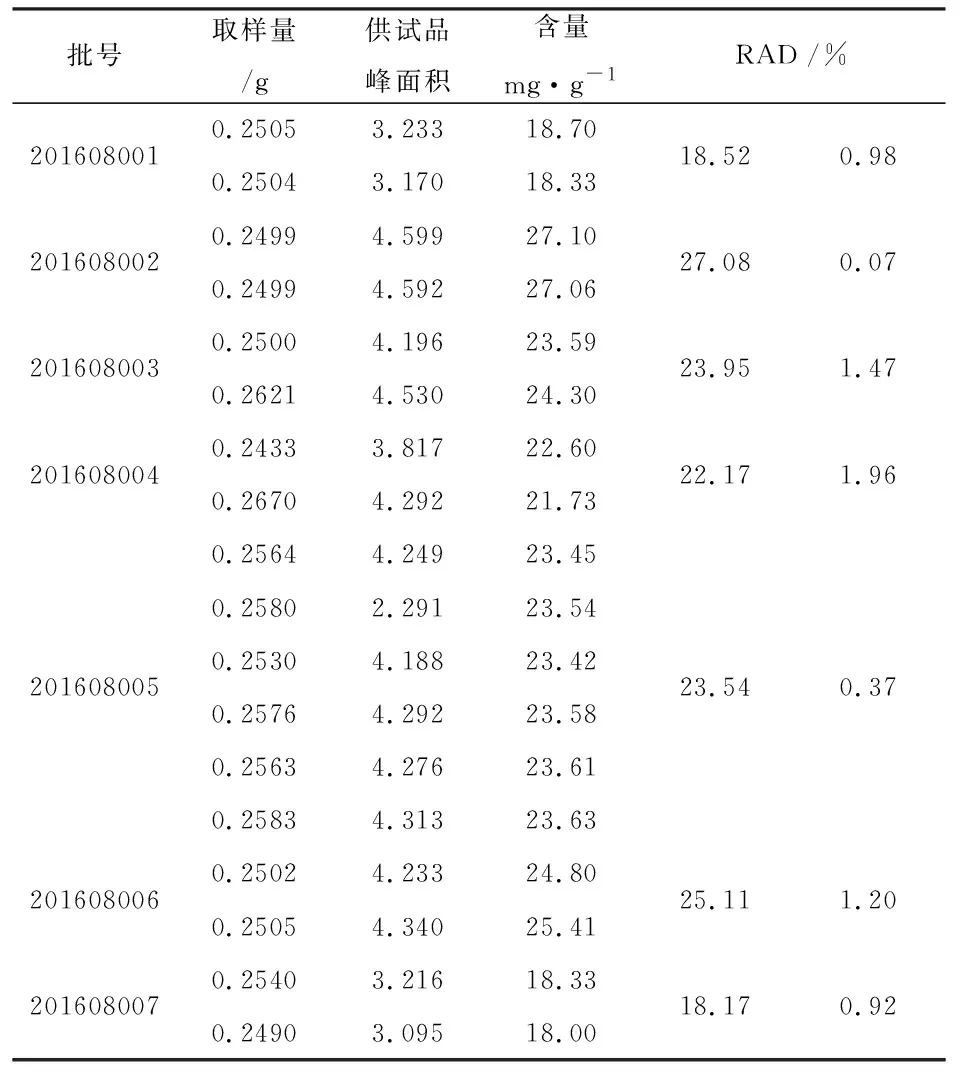
表10 样品测定试验—脯氨酸
