阿尔茨海默病及其治疗药物研究进展
2018-12-18董一筱陈乃宏
朱 杰,张 钊,董一筱,陈乃宏
(1.中国医学科学院北京协和医学院药物研究所神经科学中心,北京 100050;2.中国科学院过程工程研究所生化工程国家重点实验室,北京 100190;3.天津中医药大学药学院,天津 300193)
随着全球老龄化的进程加快,我国作为一个人口大国,罹患阿尔茨海默病(Alzheimer’s disease, AD)的老年患者激增。AD作为一种难以预防和治愈的神经退行性疾病,给老年人及其家属带来严重的经济和生活负担。然而,迄今为止,AD的发病机制众说纷纭,根据其相关发病机制研发的治疗药物在临床阶段相继宣告失败,现有的药物只能延缓AD的发病进程。因此,本文对AD的治疗现状及现有临床药物进行总结,以目前主流的AD发病机制为依据,深入探讨AD可能的发病机制,为AD相关药物的研发指明方向。
1 AD治疗现状与现有临床药物研发
AD多发于70岁以上老人,主要病理特征为脑内β淀粉样蛋白异常聚集形成的老年斑、Tau蛋白过度磷酸导致的神经元内纤维缠结、脑区不同程度萎缩。AD的致病机制非常复杂,具体机制尚不明确。目前,临床用于治疗AD的药物主要是非竞争性N-甲基-D-天冬氨酸(N-methyl-D-aspartic acid, NMDA)受体拮抗剂和胆碱酯酶抑制剂。目前认为,β淀粉样蛋白的异常堆积及Tau蛋白的过度磷酸化是AD发展的主要原因。随着针对清除β淀粉蛋白药物研发的失败,一些新的关于AD发病机制的科学研究也在开展中,并取得了相应的成果,为AD的研究及其药物的研发开辟了新的方向。
1.1临床一线治疗AD的药物早在1976年,研究发现AD患者死后脑样本中大脑皮层处的乙酰胆碱转移酶存在明显消耗的现象。Bartus等[1]随后发现,较高剂量的毒扁豆碱可以通过阻断乙酰胆碱降解的方式帮助老年人改善记忆衰退的现象,并提出AD致病的胆碱能假说。目前,FDA批准上市的5种治疗AD的药物中,多奈哌齐、利凡斯的明和加兰他敏是胆碱酯酶抑制剂(cholinesterase inhibitor, ChEI)类药[2]。这些药物只能改善AD发病早期或中期出现的认知障碍,对于晚期患者并没有治疗作用。
临床上,另一种治疗AD的药物是以美金刚为代表的NMDA受体拮抗剂类药物。该类药物可以通过抑制钙离子长时间内流超载的方式,抑制由β淀粉样蛋白引起的神经元过度兴奋,减少神经元损伤,进而起到治疗作用。虽然美金刚可以缓解AD的症状,但是临床试验证明,美金刚对中、重度AD患者疗效不确切。
1.2β淀粉样蛋白级联假说及其药物研发1992年,Hardy等[3]发现,淀粉样前体蛋白 (amyloid precursor protein,APP)基因的突变,会导致脑内β淀粉样蛋白的异常聚集,并提出了AD的β淀粉样蛋白级联假说。目前,以β淀粉样蛋白为靶点的治疗策略是:减少β淀粉样蛋白的生成、加强β淀粉样蛋白从脑内的清除、抑制β淀粉样蛋白的聚集并降低其聚集物的毒性。近年来,针对这一理论开发的药物主要是 γ-分泌酶和β-分泌酶的抑制剂,以及β淀粉样蛋白聚集抑制剂。在β淀粉蛋白级联假说致病机制的基础上,通过免疫方式定点靶向清除脑内过量的β淀粉样蛋白。
1.2.1通过活性疫苗刺激免疫系统的主动免疫 主动免疫的优点是通过短期给药刺激,产生长期的β淀粉样蛋白抗体。但是由于自身抗原的原因,易产生副作用,特别是在中老年AD患者中不良反应更多。第一代主动免疫药物AN1792可以明显性清除AD模型小鼠脑内的老年斑,并可以改善小鼠的认知功能,然而,在IIa期临床试验中有6%的患者出现脑膜炎而被迫终止。鉴于AN1792的副作用,目前的主动免疫药物主要通过靶向β淀粉样蛋白片段的方式,来降低副作用。主动免疫药物CAD-106是进入临床Ⅲ期试验阶段的唯一疫苗,并已用于AD的预防研究。目前,进行临床试验的主要主动免疫药物总结见Tab1[4-5]。
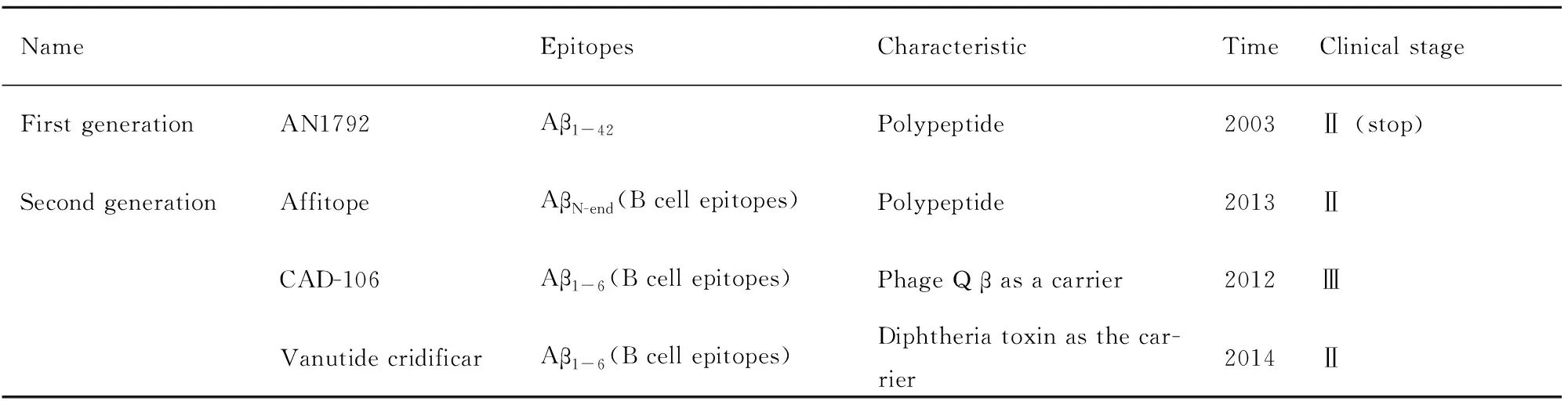
Tab 1 Major active immune drugs
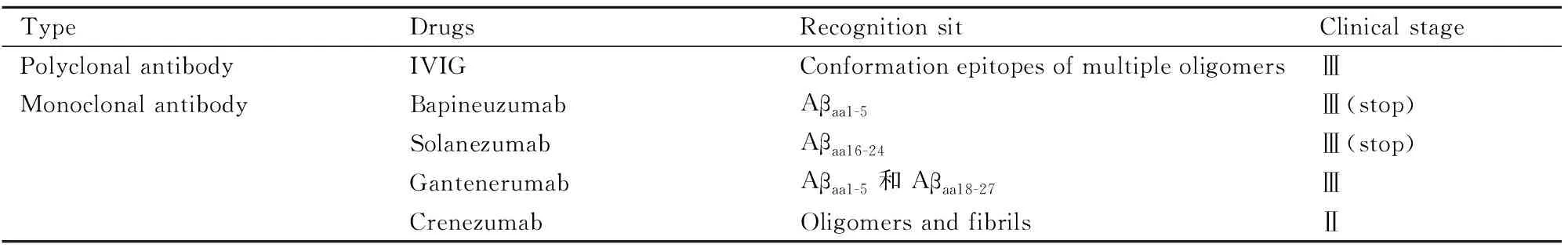
Tab 2 Major passive immune drugs
1.2.2通过外源性抗体清除β淀粉样蛋白的被动免疫 与主动免疫相比,被动免疫具有确保一致的抗体滴度和通过停止给药来控制副作用的优点。单克隆抗体(monoclonal antibodies, mAbs)的主要缺点是需要重复给药,并且药物的成本较高,每次给药的抗体用量也比较大,可能会给使用的患者造成很大的经济负担。在过去约15年的时间中,科学家们已设计了多种mAbs,用于结合和清除β淀粉样蛋白,一些药物已进入人体试验阶段(Tab 2)[4-5]。虽然mAbs在临床试验阶段的成果不尽人意,但是从这些失败的试验中获得的经验为今后AD药物的研发提供了重要线索。
1.3Tau蛋白异常聚集相关学说及其药物研发Tau蛋白是一种维持脑内神经细胞结构的微管蛋白,Tau与微管蛋白结合也会促进微管蛋白的组装和生成,并且Tau还可以支撑和维持微管蛋白的正常生理结构。AD的主要病理特征之一——脑内神经纤维缠结的主要成分即过度磷酸化的Tau,Tau的异常聚集与AD的发病呈正相关,Tau的异常聚集是额颞叶痴呆(frontotemporal dementia)和AD的病理标志[6]。当Tau过度磷酸化后,会降低其与微管蛋白的结合能力,进而导致微管蛋白形成神经纤维缠结而不能维持其正常形态。另外,磷酸化的Tau还会阻碍APP的运输,导致细胞内APP水平增加,进而导致β淀粉样蛋白的异常堆积。然而,Ittner等[7]在2016年发现,在早期AD患者中,Tau特异位点的磷酸化可以抑制β淀粉样蛋白毒性,对机体有一定保护作用。因此,Tau与β淀粉样蛋白之间存在一定的相关性,共同介导AD病程。
鉴于通过β淀粉样蛋白作为药筛靶点筛选出的治疗AD药物在临床试验阶段没有明显效果的事实,Tau作为潜在替代药物筛选靶点而备受关注。大量的实验数据也表明,Tau在AD病理过程中起到至关重要的作用。目前,旨在降低Tau蛋白磷酸化水平或防止其过度翻译聚集或控制该蛋白病理学改变的几个候选药物,正在进行临床试验[8]。
2 AD发病机制研究进展
2.1β淀粉蛋白与朊病毒的关系人们在对AD患者脑部病理切片的研究中发现,β淀粉样蛋白斑块的出现是通过相互联系的脑区逐渐扩散,而不是随着病情的发展在不同脑区随机聚集。大脑中β淀粉样蛋白的这种渐进式的传播方式证明β淀粉样蛋白的错误折叠和聚集存在自我传播的构象。Meyer-Luehmann等[9]将AD患者脑内的β淀粉样蛋白聚集体注入还未发病的过表达人源β淀粉样蛋白的年轻小鼠后,会引发小鼠内源性β淀粉样蛋白的大量沉积。Condello等[10]认为,这些实验表明β淀粉样蛋白的自体传播与慢性瓦斯病和牛海绵状脑病中发现的朊病毒的错误折叠和自体繁殖存在强烈的相关性。
目前,对于β淀粉样蛋白的传播机制尚不清楚,原β淀粉样蛋白纤维、寡聚体和纤维状态的β淀粉样蛋白可能是β淀粉样蛋白传播的潜在聚集种子。与传统朊病毒不同的是,Penke等[11]并没有发现β淀粉样蛋白、Tau、α-核突触可以从一个人传播到另一个人的依据,现在的实验结果只能表明β淀粉样蛋白有很高的朊病毒特性。
2.2基因突变致病学说从β淀粉样蛋白级联假说提出后,人们陆续发现了大量的AD易感基因。通过转基因技术开发的过表达APP/PS1的小鼠是目前AD相关药物筛选的主要动物模型。最近,大规模的全基因组关联研究(genome-wide association studies, GWAS)已经确定了20个新的AD风险的基因座。这些基因分为几个常见的途径参与AD病程,主要涉及:脂质代谢(APOE、SORL1、ABCA7、DSG2、CLU),突触功能,内吞作用(PICALM、CDZAPBIN1、SORL1),炎性反应(CD33、MEF2C、HLA-DRB5/HLA-DRB1CR1、MS4A、ABCA7、TREM2、JNPP5D、 CLUEPHA1)等。尽管确定了这些新的基因座,但是在鉴定解释与AD风险相关的功能变异体方面几乎没有进展。主要基因介绍如下:
2.2.1ABCA7 ABCA7是膜转运蛋白,位于染色体19p13.3上。ABCA7主要在小胶质细胞中表达,ABCA7中的多位点突变会导致小胶质细胞清除β淀粉样蛋白的功能丧失,进而导致AD进程中β淀粉样蛋白堆积。另外,ABCA7还会通过C1q补体途径,在调节小胶质细胞吞噬清除β淀粉样蛋白聚集体的过程中具有不可替代的作用。
2.2.2BIN 该基因编码核细胞质衔接蛋白的几种亚型,其中一种最初被鉴定为具有肿瘤抑制因子特征的MYC相互作用蛋白。在中枢神经系统中表达的BIN基因异构体可能参与突触小泡内吞,并可能与突触足蛋白、内皮细胞和网格蛋白相互作用。在肌肉中普遍表达的同种型的异构体定位于细胞质和细胞核,并激活与caspase无关的凋亡过程。BIN1特异性表达于小胶质细胞,是晚发性阿尔茨海默病(late-onset Alzheimer disease, LOAD)的遗传风险因素,其在多个GWAS研究中被确定与AD相关。BACE1主要表达于脑的神经元中,负责β淀粉样蛋白产生的1型跨膜天冬氨酰蛋白。BIN1的受损会增加细胞BACE1水平,并减少BACE1溶酶体降解,导致β淀粉样蛋白堆积。
2.2.3CD33 CD33是由脑内的神经胶质细胞和巨噬细胞特异性表达的骨髓细胞受体。CD33的过度表达与AD患者脑中的斑块增加、认知衰退和疾病的严重程度相关,并且在CD33基因敲除小鼠模型中发现,敲除CD33后可降低β淀粉样蛋白水平和减少斑块的形成。CD33位于染色体19q13.33上的CD33位点内的变体与AD相关。变体rs3865444可能介导修饰的剪接,通过影响CD33外显子II剪接,使CD33抑制胶质细胞摄取β淀粉蛋白。Malik和Jiang等[12-13]研究发现,rs12459419是与LOAD高度相关的突变基因,rs12459419可以提高BV2细胞外显子II剪接效率,促进BV2细胞的活化。两种变异体与染色质标记相关,表明在脑组织中的低/抑制性转录,但其与骨髓细胞中活性转录起始位点密切相关。
2.2.4CASS4 CASS4是CAS家族的第4位成员,目前对于CASS4与AD关系的研究较少。通过其结构功能分析表明,它们与众所周知的家族成员有很多相同结构。CASS4在免疫调节方面发挥作用,因此该基因可能与发育障碍性疾病、AD、癌症和自身免疫疾病相关。CASS4可能有保守的CAS家族细胞骨架功能,CASS4可能在轴突运输中起作用,并影响AD发病阶段β淀粉样蛋白和Tau的堆积。
2.2.5补体受体1(complement receptor 1,CR1) CR1是在包括小胶质细胞在内的与免疫相关细胞的细胞膜上表达的糖蛋白,其编码基因CR1位于染色体1q32上,由4个不同等位基因编码。该基因的大小、转录和频率在不同群体之间有变化。Fonseca等[14]报道,CR1与APOE4携带者的疾病密切相关。CR1在补体级联调节免疫激活中起主要作用,介导小胶质细胞突触的修剪。CR1的表达可以阻止小胶质细胞的活化,抑制其吞噬β淀粉样蛋白。由于CR1在脑和外周的几种免疫相关的细胞中都表达,所以目前还不清楚CR1与AD的遗传是否相关。
2.2.6Clusterin(CLU) CLU也称为apoJ,是在中枢神经系统内丰富表达的脂蛋白。它调节β淀粉样蛋白原纤维形成和毒性,并促进横跨血脑屏障的β淀粉样蛋白的转运。目前有研究表明,高脂血症可能会促进β淀粉样蛋白协同作用,促进AD的进程[15]。GWAS鉴定了CLU基因中的3个单核苷酸多态性rs11136000、rs2279590、rs9331888与白种人的AD明显相关。
2.2.7MS4A 该基因位于11q12染色体。MS4A家族内的多个基因(MS4A4A、MS4A4E、MS4A6A、MS4A6E)与AD相关,MS4A6A上游和MS4A2下游的rs983392作为AD易感基因的突变位点,与降低LOAD风险有关联。该多核苷酸突变位点与在脑组织和神经元细胞中具有低转录的染色质标记相关,并且与外周原代人单核细胞中的增强子和活性转录位点相关。另外,MS4A6E mRNA表达和rs670139与AD脑组织中的神经纤维缠结和斑块形成阶段相关。这些跨膜蛋白的功能涉及钙离子内流、调节内吞作用、信号传导,并可能作为化学感受器。
2.2.8MEF2C 该基因座对肌肉的分化增加起作用,其编码蛋白具有反式激活和DNA结合活性,可能在维持肌肉细胞的分化状态中发挥作用[16]。转录因子MEF2家族蛋白存在调节突触数和减少树突棘的功能,因此在记忆和学习中起重要作用。MEF2C被认为是APP水解加工的调节剂。MEF2C基因中常见的单核苷酸多态性(rs190982)可能与高加索人群中的LOAD有关联效应。
2.2.9NME8 虽然NME8与神经退行性疾病之间的关系尚未得到很好的报道。NM23家族成员NME8与6型原发性纤毛运动障碍相关。在脑中,由NM23家族编码的核苷二磷酸激酶(nucleoside diphosphate kinase,NDPK)涉及调节神经细胞增殖、分化和神经突起生长。虽然NDPK在神经退行性疾病中的作用尚未完全报道,但AD中NDPK的蛋白表达水平中度降低会影响神经元功能,如神经突起增生和轴突发芽通过改变细胞骨架蛋白的表达,会引起脑内神经细胞的异常增殖和分化。此外,由nm34-H2基因编码的nm23核苷二磷酸激酶/转移抑制蛋白(PuF)可调节APP基因的启动子,这可能会通过干扰近端调节元件上PuF的方式,介导AD进程。Liu等[17]在认知研究的实验中发现,健康个体NME8的3种基因型的临床痴呆评分量表评分不同,两年后差异更大,GG等位基因的存在有利于提高认知。因此,NME8 rs2718058可能会改善一般人群的认知,甚至对于AD的临床前阶段出现的认知障碍也有一定作用。
2.2.10PTK2B 该基因编码细胞质蛋白酪氨酸激酶,其参与钙诱导的离子通道调节和激活图谱激酶信号通路。编码的蛋白质可以代表增加钙通量的神经肽激活受体或神经递质之间的重要信号中间体,以及调节神经元活性的下游信号。编码的蛋白质响应于细胞内钙浓度、烟碱乙酰胆碱受体激活、膜去极化或蛋白激酶C活化的增加,而经历快速的酪氨酸磷酸化和活化。Dourlen等[18]发现,磷酸化的蛋白酪氨酸激酶与AD患者及转基因Tau鼠脑中磷酸化的Tau共定位,参与Tau的磷酸化和聚集等过程。这些数据表明,PTK2B作为Tau毒性的早期标记物和体内调节剂。综合现有的数据,PTK2B可能是Tau磷酸化介导的AD发病机制中的主要参与者。
2.2.11ZCWP1 最近,Efthymiou等[19]进行的GWAS已经确定了ZCWPW1中的新型变体rs1476679与白种人和北韩汉族人群LOAD强烈相关。此外,ZCWPW1基因座内的PILRB和GATS表达水平也与AD病程相关。
2.3小胶质细胞介导的炎症对AD的影响神经炎症是神经细胞坏死的主要诱因之一。神经炎症主要是由小胶质细胞介导的,血管周围髓细胞和星形胶质细胞对其有一定的辅助作用。神经炎症作为AD的一种触发机制,与β淀粉样蛋白的堆积也有一定的联系。小胶质细胞在免疫防御中发挥巨噬细胞的作用,小胶质细胞可以吞噬APP/PS1模型鼠脑内的β淀粉样蛋白。小胶质细胞对β淀粉样蛋白的清除作用是通过TLR4受体介导,β淀粉样蛋白同样也是TLR4的配体,因此,脑内β淀粉样蛋白的聚集可能会导致TLR4的慢性暴露,进而导致TLR信号传导功能障碍和炎性反应。Hong等[20]的最新实验结果表明,在早期AD小鼠模型中,过度活化的小胶质细胞在清除β淀粉样蛋白的同时,也会过度修剪突触,造成突触丢失。
小胶质细胞在AD的发展过程中存在双重作用:一方面,小胶质细胞通过清除脑内多余β淀粉样蛋白,可以减轻AD进程中出现的认知障碍;另外,小胶质细胞的过度活化,会增加如TNF-α、IL-1β等促炎因子的释放,反而对AD的发展起到促进作用[21]。因此,以小胶质细胞的炎症释放通路作为药物研究靶点,开发选择性切断小胶质细胞释放促炎因子的药物,可能是减轻AD脑内神经炎症的一种方法。
2.4神经中枢和昼夜节律对AD的影响及其治疗生物钟是生物体内分子和细胞震荡构成的跟踪时间的内部时钟。Hut等[22]的研究发现,不论是果蝇还是人类,生物钟都控制着生命体的日常生命活动。生物体在睡眠、饮食、生殖、交配、躲避危险等生理活动中始终处于最佳状态,是在生物钟的控制下完成的。日常昼夜节律在调节机体健康中存在重要作用,昼夜节律失常往往是一些代谢疾病、癌症、血管性疾病的诱因。越来越多的证据表明,睡眠障碍可能更早于AD早期出现的认知功能障碍。
目前,对于睡眠和昼夜节律失常的治疗主要通过改善睡眠质量和正常化睡眠的治疗方法。最近,经美国FDA批准的可以有效治疗失眠症的食欲素受体拮抗剂Suvorexant,可以为今后研究食欲肽与AD的关系创造可能。昼夜节律与AD的关系正处于临床前发展阶段,通过改善睡眠质量和昼夜节律时间,可以对AD患者及其护理人员的生活质量产生直接的积极影响。在生命早期优化睡眠质量和昼夜节律可能是预防或延缓AD发展的手段,对于生物钟方向的研究可能成为今后治疗神经退行性疾病的一个独特方式。
2.5γ电位震荡与AD的关系虽然研究已经证明,多种神经系统疾病都会破坏γ震荡的平衡,然而,γ震荡与细胞病理之间的关系尚不清楚。Iaccarino等[23]发现,通过40赫兹γ射线的LED灯照射AD模型小鼠,可以降低脑内Aβ1-40和Aβ1-42水平,减轻小鼠脑内老年斑,并且该射线在Tau蛋白过表达的小鼠模型中也可以降低Tau的水平。
特定的γ射线可以诱导小胶质细胞形态转化的有关基因,增加小胶质细胞对β淀粉样蛋白的清除速率,增加其对β淀粉样蛋白的内吞,进而减少β淀粉样蛋白的沉积。这种治疗方式跟传统的AD疗法存在本质的区别:Canter等[24]认为,γ射线是对脑内的系统治疗,恢复脑内正常的神经回路。目前,对于γ射线的研究还处于动物模型阶段,对人类是否有效还有待科学的验证。
2.6外泌体与AD早期诊断外泌体存在于身体各个部位,它是体内大多数细胞分泌的纳米大小(40~1 000 nm)的细胞外囊泡。外泌体在过去几年中一直被认为是细胞排放垃圾的一种形式,现在外泌体被证明是重要的细胞间信使,对健康和疾病有重要的作用。外泌体在AD进程中存在多方面作用,外泌体携带的致病蛋白有助于疾病在脑内的扩散,并且外泌体可以诱导星形胶质细胞的凋亡,间接损害神经元的正常功能。另一方面,外泌体可以降低细胞外淀粉样蛋白的负荷,并且可以作为AD早期诊断的标志物。
3 AD研究主要趋势
3.1β淀粉样蛋白级联假说面临挑战25年前,人们就发现APP的突变与AD的发展存在密切关系,并最终建立了β淀粉样蛋白假说。另外,大量的实验数据和动物模型都可以证明这一假说的成立。该假说已经是AD发病机制的主导假说,以及绝大多数实验治疗方法的基础。然而,临床上抗淀粉样蛋白的药物研发的失败,总结出了β淀粉样蛋白的产生和堆积只是AD进展过程中主要的病理特性,但不是其致病因子。另外,越来越多的研究发现,APP、β淀粉样蛋白、γ分泌酶、β分泌酶对机体也有一定的保护作用,以其作为治疗靶点可能是药物研发失败的原因。因此,β淀粉样蛋白级联假说在得到综合实验结果支持的同时,也要面对新的挑战。
3.2AD易感基因是AD发病机制的突破口AD的精确分子发病机制尚不明确,高、低表达的基因,以及表观遗传学和环境因子参与了AD的发展。此外,脑外伤、脑缺血、缺血/再灌注、β淀粉样蛋白清除受阻,都会对蛋白质异常平衡和线粒体功能障碍有影响。
因此,要探索AD的起始和起源问题,就要搞清楚衰老中相关蛋白的相互作用失调和不平衡的原因。蛋白的过度表达和减少很可能是其相关基因的突变介导的。目前,人们已经发现,APP、PSEN1、PSEN2、ApoE、Tau等基因的突变,都会导致β淀粉样蛋白的堆积。最近,GWAS发现的AD易感基因对于探索AD的发病机制存在很大的研究意义。因此,基于基因水平对AD的研究,从遗传因素和防御机制两个方面出发,可能是AD治疗的突破口。
3.3构建新型的AD药物筛选模型AD是一种复杂的神经退行性疾病,受机体和环境各个方面的影响。传统的AD细胞模型和动物模型只能单一地表现AD的1个或几个症状,并不能代表AD患者的所有病程。通过APP/PS1小鼠模型筛选的治疗AD的药物在动物模型上存在很好的疗效,然而,在临床试验阶段发现对AD患者并没有治疗作用。鉴于无法创造良好的AD模型,一部分科学家转向了神经环路调控的方向,以期通过改善神经环路障碍治愈AD。因此,开发能够完全模拟AD患者病理过程的动物模型在AD药物筛选中至关重要。
现有的AD动物模型主要是啮齿类大鼠或小鼠模型。但是AD涉及的是较高的认知功能损害,不管是啮齿类动物,还是兔子和犬,都不能完全模拟。因此,通过对非人灵长类动物的研究,基于人类与非人灵长类动物的大脑在功能网络的整体结构和组成方面的相似性,开发相应的AD动物模型,可以克服现有动物模型不能完全模拟AD患者所有病程的困境。
3.4早期诊断可能是AD药物研发的主要方向在过去的几十年中,超过50个候选药物顺利通过Ⅱ期临床试验,但均在Ⅲ期试验中失败。大多数的候选药物都是以不同形式的β淀粉样蛋白作为靶点,以期清除斑块或组织淀粉样蛋白的异常聚集。这些试验的失败在改变人们对AD认识的同时,也改变了AD药物的研究目的。既然我们不能让受损的神经突触恢复原样,那么我们下一步研发的方向就应该是早期诊断。
4 小结
综上所述,人口老龄化是全世界无法回避的事实, AD等老年性疾病引发的社会问题已然非常严峻,但合理有效的临床药物依然空缺。近期多项AD临床试验失败的部分原因是由于在AD的晚期阶段,药物干预不能逆转神经突触的修复。另一方面,鉴于AD的多靶点性,AD的机制目前尚不明确,现有的模拟AD的动物模型尚不完善,AD药物研发近年来一直停滞不前。在度过很短的茫然期后,对于AD基础研究空前高涨: AD 易感基因的预测,早期标志物的寻找,神经环路的研究,生活习惯、饮食、休息节律等的调控,阮病毒样传播方式的求证,外泌体的研究,新的动物模型的寻找,甚至光刺激的参与等,都取得了一定的进展。AD 作为复杂的神经退行性疾病,其本身体现的即为一个多靶点效应。因此,按照现在单一靶点治疗的模式可能事倍功半,今后对于AD的药物研发的方向必然是多学科的结合和交流才能产生突破性的进展。
