制备液相色谱法快速分离制备硫酸依替米星杂质G
2018-12-18毛菊红金坚王晓霞
毛菊红,金坚,王晓霞
制备液相色谱法快速分离制备硫酸依替米星杂质G
毛菊红1,金坚1,王晓霞2
(1.江南大学药学院,江苏 无锡 214122;2.无锡济民可信山禾药业股份有限公司,江苏 无锡 214028)
为快速获得硫酸依替米星杂质G样品,使用制备液相色谱分离制备硫酸依替米星杂质G。具体方法为,将硫酸依替米星样品用水溶解上样于大孔树脂柱,再使用硅胶柱粗分处理后,用制备液相色谱法快速制备分离;制备色谱柱为XBridgeTMPrep C185μm OBDTM(19 mm×100 mm,Column),流动相A为水-氨水-冰醋酸(96∶3.6∶0.4,∶∶),流动相B为甲醇,梯度洗脱,快速制备杂质G样品。采用高效液相-离子色谱、核磁共振(NMR)、电喷雾电离质谱(ESI-MS)对制备后的杂质G样品进行结构确证。样品经大孔树脂柱、硅胶柱粗分,再利用制备液相色谱从硫酸依替米星原料药中成功分离制备杂质G样品,纯度为96.02%,经结构确证为6″-羟基依替米星,为合成的副产物。建立制备液相快速分离硫酸依替米星杂质G的方法,为将来开展杂质G研究提供了参考。
硫酸依替米星;结构鉴定;制备液相;杂质G
硫酸依替米星( Etimicin sulfate) 是我国科技人员研究开发的具有自主知识产权的一类新药,具有良好的抗感染治疗效果,具有抗菌活性强,抗菌谱广,耐受性好的特点[1]。硫酸依替米星属氨基糖苷类抗生素,不良反应主要为耳毒性、肾毒性等,但发生率大都在1%以下,低于阿米卡星、妥布霉素和头孢呋辛等其他同类产品[2]。硫酸依替米星现收载于《中国药典》2015年版二部[3],特定杂质结构如图1所示。硫酸依替米星特定杂质结构如表1所示。

图1 特定杂质结构
表1 硫酸依替米星特定杂质结构表
杂质编号名称A环B环C环 R1R2R3R4R5 依替米星NH2HNH2HC2H5+ A1-N-乙基-加洛糖胺脱去A环HC2H5+ B1,3-N-二乙基-加洛糖胺脱去A环C2H5C2H5+ C NH2HNH2HC2H5脱去C环 D西索米星*NH2HNH2HH+ E庆大霉素C1aNH2HNH2HH+ F小诺霉素 CH3NHHNH2HH+ G OHHNH2HC2H5+ H奈替米星*NH2HNH2HC2H5+ I1-N-乙基小诺霉素CH3NH HNH2HC2H5+ J6’-N-乙基庆大霉素 C1aC2H5NHHNH2HH+ K3-N-乙基依替米星NH2HNH2C2H5C2H5+ L中间体P1CH3CONH HCH3CONHCH3COH+
表1中,*表示4′,5′之间为双键;杂质D(西索米星)、杂质E(庆大霉素C1a)、杂质F(小诺霉素)、杂质H(奈替米星)有市售外,其余杂质均暂无市售。
本研究建立了制备液相色谱法制备硫酸依替米星中杂质G(依替米星有关物质)的方法,制备获得纯度95%以上的杂质G样品,用于硫酸依替米星质量研究并辅助指导工艺研究。
1 材料
1.1 仪器
本研究所用仪器主要有2545制备液相色谱仪(美国Waters)、2424蒸发光检测器(美国Waters)、ICS-5000离子色谱仪(美国赛默飞世尔)、积分脉冲安培电化学检测器(美国赛默飞世尔)、MILLI-Q去离子水发生器(美国密理博公司)、AVANCE 600核磁共振谱仪(德国Bruker)、1100 LC/MSD Trap液质联用仪(美国Agilent)。
1.2 试药与样品
试药与样品为:硫酸依替米星(批号160902-Y,无锡济民可信山禾药业股份有限公司),层析1号树脂(上海华震科技股份有限公司),100-200硅胶(青岛海洋),HPLC级甲醇(默克,批号为I0910907738,纯度≥99.9%),HPLC级冰醋酸(TEDIA,批号为16090277,纯度≥99.7%),乙醇、氨水均为分析纯。
2 方法与结果
2.1 制备液相色谱条件
色谱柱:XBridgeTMPrep C185μm OBDTM(19 mm×100 mm,Column)。流动相:A相,水-氨水-冰醋酸(96∶3.6∶0.4,∶∶);B相,甲醇,梯度洗脱。流速:20 mL/min。柱温:40~50 ℃。进样量:1 mL。ELSD参数:载气压力0.3~0.4 MPa。Gain:10.流动相梯度如表2所示。
表2 流动相梯度
时间/min 流动相A/(%) 流动相B/(%) 08020 57525 127525 204060 251090 301090
2.2 离子色谱检测条件
本研究检测方法采用《中国药典》2015年版二部硫酸依替米星标准含量测定第一法[3],积分脉冲安倍检测器(PAD)较蒸发光检测器(ELSD)灵敏度更高[4],更适合对氨基糖苷类抗生素组分/杂质控制进行测定。色谱柱:月旭(Welch ultimatelp)-C18十八烷基硅烷键合硅胶填充柱(4.6 mm×250 mm,5 μm);流动相:0.2 mol/L三氟乙酸(含0.05%的五氟丙酸,1.5 g/L的无水硫酸钠,0.8%(V/V)的50%氢氧化钠溶液,用50%氢氧化钠溶液调节pH值至3.5)-乙腈(96∶4);柱温:35 ℃;流速:1.0 mL/min;检测器:积分脉冲安培电化学检测器;检测电极:金电极(直径为3 mm);参比电极:Ag/AgCl复合电极;柱后加碱(50%氢氧化钠溶液1→25,流速为0.5 mL/min);检测波形:四电位波型。检测电位设定如表3所示。
2.3 杂质制备
2.3.1 杂质初步分离
取硫酸依替米星用水稀释后先上样于大孔树脂柱,用不同浓度乙醇洗脱。收集洗脱液,再经硅胶柱分离,作为供试品。将供试品浓缩去除溶剂后,用适量超纯水稀释,备用。
2.3.2 进一步制备分离
将初步分离后的杂质样品在制备液相上进样1 mL,按制备液相色谱条件制备。观察图谱,有峰出现后,开始收集样品至出峰结束,按出峰顺序分别标注样1、样2、样3、样4、样5,如图2所示。
将收集的样品按离子色谱条件检测,采用面积归一法,计算杂质纯度。如果收集样品检测纯度小于95%,则将制备后的收集样品再继续制备,直至纯度大于95%.
表3 检测电位
时间/s电位/V积分 0.00+0.10 0.20+0.10开始 0.40+0.10结束 0.41﹣0.20 0.42﹣0.20 0.43+0.60 0.44﹣0.10 0.50﹣0.10

图2 制备液相第一次制备
硫酸依替米星HPLC-PED检测出峰时间为26 min,杂质G相对于硫酸依替米星的相对保留时间为1.12,因此杂质G出峰时间约为29.12 min(26 min×1.12)。
将样1、样2、样3、样4、样5分别经离子色谱法检测,根据出峰时间可判定为样3中含有杂质G,出峰时间为29.184 min,如图3所示;纯度为21.88%,如表4所示。
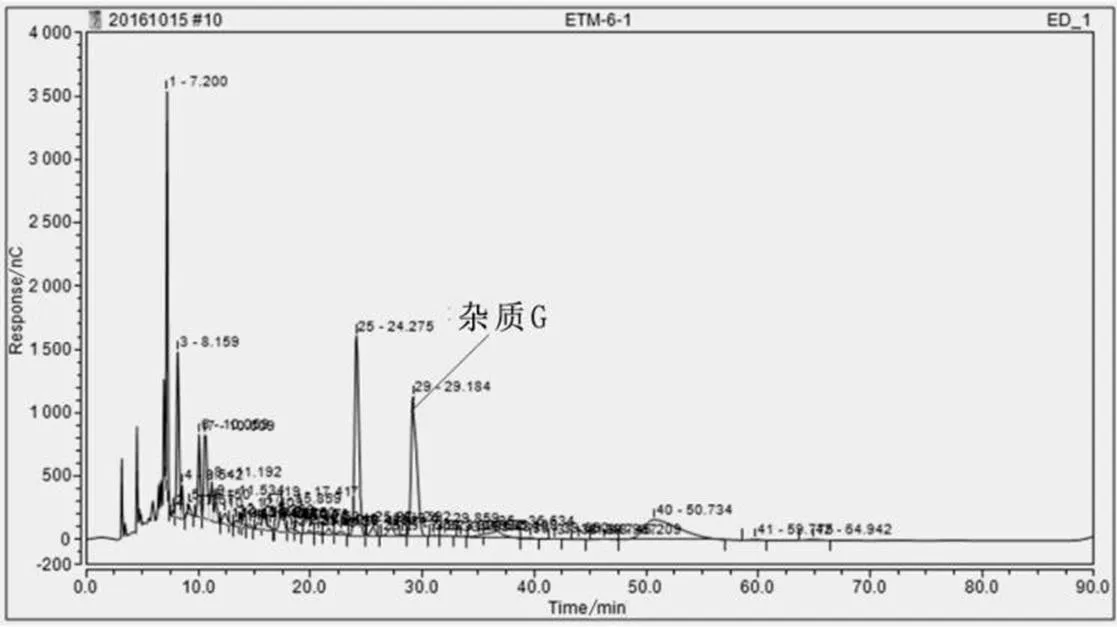
图3 杂质G检测图谱
将样3继续按制备液相色谱条件制备,按离子色谱条件检测样品纯度。第五次制备图谱如图4所示,样5-2为杂质G(出峰时间为29.155 min),检测图谱如图5所示,样品纯度为96.02%,检测数据如表5所示。表5中参考值为庆大霉素C1a的氢谱和碳谱数据。
表4 杂质G纯度
出峰序号保留时间/min面积/nC·min峰高/nC相对面积/(%)相对峰高/(%) 17.200401.7983 144.1649.9229.32 27.76710.12957.0460.250.53 38.159304.4941 306.0087.5212.18 48.54247.314265.3731.172.47 59.15017.93580.5190.440.75 610.059138.367650.9993.426.07 710.609243.971663.0446.026.18 811.19280.156307.9941.982.87 911.53450.417175.6391.241.64 1012.40940.00489.3690.990.83 1112.7176.51727.3740.160.26 1213.35016.44753.7250.410.50 1313.6928.21134.1800.200.32 1414.0008.11827.8900.200.26 1514.57513.61034.5400.340.32 1615.35021.13256.9070.520.53 1715.85964.132168.6331.581.57 1816.3504.15918.9480.100.18 1917.417102.191243.9452.522.28 2018.27517.69838.6860.440.36 2118.7177.96421.8490.200.20 2219.90911.57933.0760.290.31 2320.7009.91023.2500.240.22 2421.65922.37851.7190.550.48 2522.975701.5041 594.98117.3214.87 2624.45924.36933.2150.600.31 2725.59232.55273.4990.800.69 2826.3593.0609.5740.080.09 2929.184886.1681 100.02321.8810.26 3029.85940.88567.1981.010.63 3131.0752.0774.6940.050.04 3232.0922.9814.2170.070.04 3333.4841.6323.1930.040.03 3435.10014.50817.2360.360.16 3536.63482.35654.6962.030.51 3639.6505.5256.5590.140.06 3741.8171.1202.0760.030.02 3843.9671.2981.8600.030.02 3946.2096.1315.2100.150.05 4050.734581.242157.77414.351.47 4159.7755.5955.2750.140.05 4264.9428.3176.4330.210.06 总计: 4 049.95110 722.592100.00100.00
图4 制备液相第五次制备

图5 杂质G(第五次制备)检测图谱
表5 杂质G(第五次制备)纯度
出峰序号保留时间/min面积/nC·min峰高/nC相对面积/(%)相对峰高/(%) 16.5500.1410.3360.010.00 26.9171.7353.5920.100.01 38.9341.0952.7060.060.01 49.1596.75111.6840.400.03 510.30032.44255.9931.910.16 613.7343.8644.7350.230.01 714.6590.8000.8930.050.00 815.2847.0056.8360.410.02 918.12513.8159.7040.810.03 1029.1551 630.031252.61096.020.72 Total 1 697.678349.089100.001.00
3 结构确证
3.1 核磁共振谱仪测定
将已制备的5-2样品(纯度为96.02%),经核磁共振谱仪测定,测试结果如表6所示。氢谱的低场区域出现化学位移分别为4.98 Hz/MHz和5.15 Hz/MHz的双重峰,可能为糖的端基氢,提示该化合物中含有2个糖。化学位移在 2.54 Hz/MHz处有一个3个氢的单峰,可能为氨基上的取代甲基。低场区域在1.22 Hz/MHz处有一个3个氢的单峰,表明存在一个连在季碳上的甲基。在1.08 Hz/MHz处有一个3个氢的三重峰,表明存在乙基。
氢谱数据与庆大霉素C1a的文献值[5]比较,发现A环6″位上的氢向低场位移到3.63 Hz/MHz和3.53 Hz/MHz,提示A环6″位上的氨基有可能被羟基取代。
与文献中庆大霉素C1a的碳谱数据进行比较,发现多出两个碳信号,化学位移为42.52 Hz/MHz和15.74 Hz/MHz,可能为连在氨基上的乙基。此外,B环1位碳信号向低场位移6 Hz/MHz,2位和6位碳信号分别向高场位移3 Hz/MHz和1 Hz/MHz,说明乙基取代在1位氨基上。A环6″碳向低场位移到66.3 Hz/MHz,结合氢谱数据,说明B环6″上的氨基被羟基所取代,其余数据与文献值基本一致。进一步分析该化合物的偶合常数,A环2″位氢的偶合常数为11.7 Hz和4.0 Hz,说明2″位氢在竖键上,A环端基氢的偶合常数为3.4 Hz,说明端基氢在横键上。A环4″位竖键氢为dq峰,其偶合常数为3.6 Hz和12.1 Hz,可推知5″位氢在竖键。C环3′位氢的偶合常数为10.7 Hz,因此2′和3′位的氢都在竖键,1′和2′位的偶合常数为4.0 Hz,因此1′位氢在横键。B环上4,5,6位氢的偶合常数都为9.2 Hz,说明1,3,4,5,6位的氢都在竖键上,与庆大霉素C1a的相对构型一致。
H-H COSY谱分析得到的自旋偶合体系信息与所推断的结构一致,相关信号如图6所示。在HMBC谱中,可观察到乙基上的氢与B环1位碳有相关,说明乙基取代在1位氨基上。A环的端基氢与B环4位碳有相关,说明A环通过糖苷键连接在B环的4位。C环的端基氢与B环6位碳有相关,说明C环通过糖苷键连接在B环的6位。A环羟甲基上的氢与A环4″,5″位碳相关,说明羟甲基连接在5″位。化学位移为1.22 Hz/MHz的甲基氢与C环3′,4′,5′碳相关,表明该甲基连接在4′位。连接在氨基上的甲基氢(δ2.54 Hz/MHz)与C环3′位碳有相关,表明甲氨基连接在C环3′位。主要HMBC相关如图7所示。
ROESY谱得到的相对构型与分析氢谱偶合常数得到的结果一致,主要ROESY相关如图8所示。鉴定杂质G的相对构型如图9所示。
表6 样品的H-NMR,C-NMR数据
序号1H-NMR(300MHz,D2O)13C-NMR(75 MHz,D2O) 实测值文献值[5]实测值文献值[5] 12.75~2.82(1H,m)2.79~2.95(1H,m)59.2651.7 22.18(1H,dt,13.0,3.6 Hz,H-e)1.09(1H,q,13.0Hz,H-a)1.96(1H,dt,12.9,4.0Hz)1.17-1.30(1H,m)34.4536.7 32.79~2.87(1H,m)2.79-2.95(1H,m)51.7250.6 43.33(1H,t,9.2Hz)3.31(1H,t,9.4Hz)89.4588.3 53.63(1H,t,9.2Hz)3.59(1H,t,9.4Hz)76.7975.4 63.34(1H,t,9.2Hz)3.25(1H,t,9.4Hz)87.8287.8 72.49(1H,dq,11.2,7.2 Hz)2.75(1H,dq,11.2,7.2 Hz) 42.52 81.08(3H,t,7.1Hz) 15.74 1′4.98(1H,d,4.0Hz)5.08 (1H,d,4.0Hz)103.39101.3 2′3.82(1H,dd,10.7,4.0 Hz)3.80(1H,dd,10.6,4.0 Hz)71.5670.2 3′2.59(1H,d,10.7Hz)2.57(1H,d,10.6Hz)65.6064.4 4′ 74.5773.3 5′4.02(1H,d,12.7Hz, H-a)3.33(1H,d,12.7Hz, H-e)4.04 (1H,d,12.4 Hz)3.31 (1H,d,12.4 Hz)70.1368.7 6′1.22(3H,s)1.20(3H,s)23.8323.0 7′2.54(1H,s)2.50(3H,s)38.9938.0 1″5.15(1H,d,3.4Hz)5.14(1H,d,3.5Hz)103.25102.2 2″2.90(1H,dt,11.7,4.0 Hz)2.81(1H,dd,3.5,7.8 Hz)52.0351.0 3″1.66(1H,dq,3.6,12.5 Hz,H-a)1.75~1.82(1H,m,H-e)1.55-1.83(2H,m)27.9727.1 4″1.45(1H,dq,3.6,12.1 Hz,H-a)1.67~1.72(1H,m, H-e)1.40-1.68(2H,m)28.0728.5 5″3.96~4.00(1H,m)3.83-3.94(1H,m)72.3171.5 6″3.63(1H,dd,12.0,3.8 Hz)3.53(1H,dd,12.0,6.8 Hz)2.57(2H,dd,10.7,6.6 Hz)66.3446.1
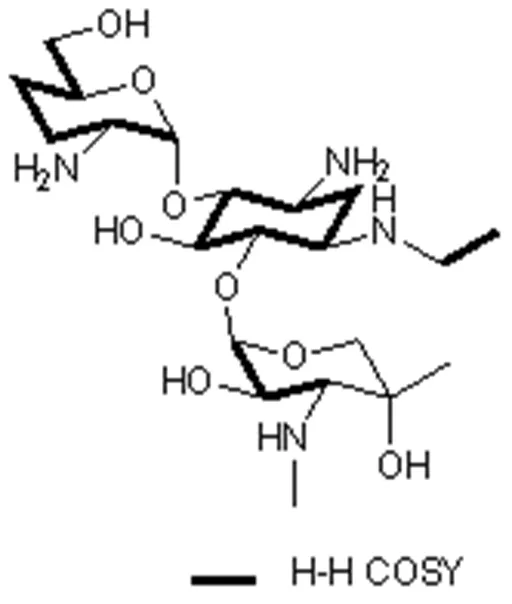
图6 杂质G的H-H COSY相关示意图
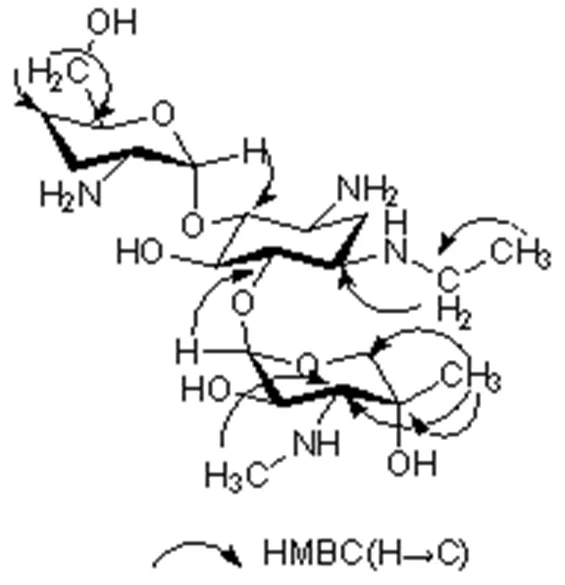
图7 杂质G的主要HMBC相关示意图

图8 杂质G的主要ROESY相关示意图

图9 杂质G的结构式
3.2 质谱解析(ESI-MS)
ESI-MSn测定数据及解析结果:①ESI-MS m/e 479.3 [M+H]+;②ESI-MS2 m/e(479.3)分别为350,344,320, 215,191,160.离子源为电喷雾质谱,正离子模式主要给出样品的分子离子加氢峰。正离子模式给出的二级质谱的裂解途径如图10所示。以上ESI-MS2信息,证明样品与推断结构相符。
4 结论
经制备液相色谱制备、离子色谱检测,可初步判定分离所得的样品为杂质G,纯度96.21%.综合NMR和ESI-MS解析,进一步确定该样品为依替米星有关物质6″-羟基依替米星,为依替米星类似物合成的副产物。
本研究仅为高纯度杂质G的制备获取,也为将来开展杂质G研究提供高纯度样品。
5 讨论
药品在临床使用中产生的不良反应除了与药品本身的药理活性有关外,有时与药品中存在的杂质也有很大关系,因此杂质的研究也是药品研发的一项重要内容。且按药品标准物质原料申报备案办法,作为有关物质检查用纯度需达到95%以上。
硫酸依替米星为半合成氨基糖苷类抗生素,合成工艺的参数变化甚至起始物料的变化,都将导致成品中杂质的变化,总体来说杂质来源复杂。袁耀佐、张玫等建立了高效液相色谱-电喷雾-离子阱质谱法推定硫酸依替米星中有关物质的结构,通过该方法检出了18种有关物质,对其中13种物质的结构进行了推定[6];李正义、晏培培等通过在色谱柱与检测器之间连接1 个分流装置,按照5∶1 分流比使少量流出液进入检测器,其余流出液被收集合并。按此方法制备出4种高纯度的杂质,分别为1-N-乙基-加洛糖胺、1-N-乙基-3′,4′-二-脱氧新霉胺、1-N-乙基-3″-N-脱甲基庆大霉素C1a、1-N-乙基小诺霉素[7],该方法虽成功分离并鉴定了杂质,但分离的4个杂质纯度未有数据。据现有技术检索,暂未有高纯度硫酸依替米星杂质G的分离纯化或制备报道。
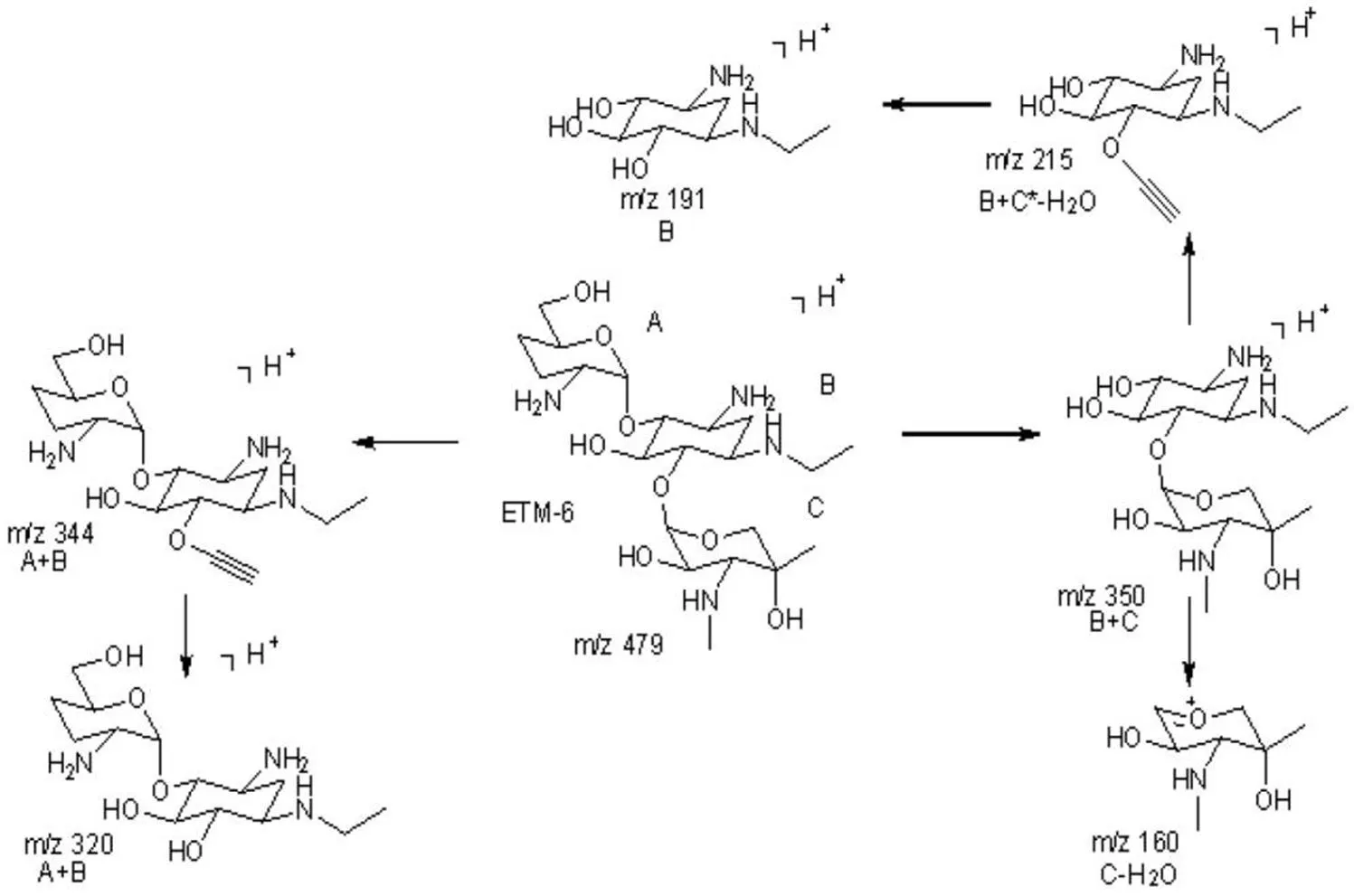
图10 杂质G的质谱裂解途径
制备液相色谱利用混合物中各组分物理化学性质的差异,使它们以不同程度分布在两个不相溶的相中,从而达到分离目的[8]。本研究使用制备液相分离制备技术从硫酸依替米星原料中分离制备了杂质G依替米星有关物质6″-羟基依替米星,首次将制备液相分离纯化技术应用于硫酸依替米星杂质制备纯化,获得纯度96.21%的杂质G样品,可用于硫酸依替米星质量标准研究或进一步开展杂质研究。
[1]胡伶俐.硫酸依替米星抗感染的临床药理探讨[J].中国医药指南,2013,11(7):171-172.
[2]陈永法,曹文帅.硫酸依替米星临床安全性研究综述[J].中国药物经济学,2012(6):41-44.
[3]国家药典委员会.中华人民共和国药典二部[S].2015版.北京:中国医药科技出版社,2015.
[4]胡昌勤.化学药品杂质谱控制的现状与展望[J].中国新药杂志,2015,24(15):1727-1734.
[5]DANIELS P J L,CHARLES L,NAGABHUSHAN T L.The gentamicin antibiotics:6-gentamicin C2b,an aminoglycoside antibiotic produced by Micromonospora purpurca mutant JI-33[J].J Antibiot,1975,28(1):35-41.
[6]袁耀佐,张玫,钱文,等.高效液相色谱-电喷雾-离子阱质谱法推定硫酸依替米星中有关物质的结构[J].分析化学,2010,38(6):817-822.
[7]李正义,晏培培,殷晓伟,等.依替米星中杂质的分离纯化及结构鉴定[J].药物分析杂志,2017,37(11):1978-1985.
[8]陈鸳谊,李行诺,张翠萍,等.高效制备液相色谱在天然产物分离中的应用[J].药学进展,2010,34(8):337-343.
2095-6835(2018)23-0047-06
C829.2
A
10.15913/j.cnki.kjycx.2018.23.047
毛菊红(1984—),女,江苏苏州人,在职研究生,助理工程师,研究方向为制药工程。
〔编辑:严丽琴〕
