10-去乙酰巴卡亭III-10-β-O-乙酰转移酶迭代饱和突变与活性位点分析
2018-12-11张育楠陈天娇朱平
张育楠,陈天娇,朱平
10-去乙酰巴卡亭III-10-β--乙酰转移酶迭代饱和突变与活性位点分析
张育楠,陈天娇,朱平
100050 北京,中国医学科学院北京协和医学院药物研究所天然药物活性物质与功能国家重点实验室/国家卫生健康委员会天然产物生物合成重点实验室
利用迭代饱和突变技术进一步提高10-去乙酰巴卡亭 III-10-β--乙酰转移酶双突变体DBATG38R/F301V的活性;通过丙氨酸扫描确定DBAT 活性中心的重要位点。
利用迭代饱和突变技术在现有双突变体 DBATG38R/F301V的基础上对 Ser396进行半饱和突变,筛选对10-去乙酰紫杉醇(DT)催化活性提高的突变体;对获得的高活性突变体蛋白进行催化 DT 的最适温度和最适 pH 测定;在最适 pH 条件下,将其分别用最适反应温度和 0 ℃温育 6 h,考察该蛋白的热稳定性;采用生物信息学方法分析活性提高的分子机制。对 DBAT 底物结合区域尚未进行分析的几个氨基酸进行丙氨酸扫描研究,分析这些位点在 DBAT 催化 10-DAB 和 DT 过程中的作用。
获得了三突变体 DBATG38R/F301V/S396I,其催化 DT 的活性为 DBATG38R/F301V的 1.6 倍,该蛋白催化DT 的最适条件为 37.5 ℃,pH 7.5;该蛋白在 37.5 ℃静置 6 h 后催化 DT 的活力为初始值的 30%,在 0 ℃静置 6 h 后为初始值的 70%;丙氨酸扫描结果显示,Asn45在 DBAT 催化 10-DAB 和 DT 时都非常重要,而 Asn42、Glu350、Glu355和Lys389位点对催化 10-DAB 的影响更大。
将活性口袋内的亲水性氨基酸向疏水性氨基酸突变可能有利于 DBAT 对 DT 的催化,但氨基酸侧链的长度和空间构型也是影响催化活性的重要因素;发现 Asn45可能是一个潜在的 DBAT 催化或底物结合位点。
10-去乙酰巴卡亭III-10-β--乙酰转移酶; 迭代饱和突变; 高活性突变体; 丙氨酸扫描; 潜在活性位点
10-去乙酰巴卡亭 III-10-β--乙酰转移酶(10-deacetylbaccatin III-10-β--acetyltransferase,DBAT)催化紫杉醇生物合成途径中的 10-去乙酰巴卡亭 III(10-deacetylbaccatin III,10-DAB)形成重要中间产物巴卡亭 III(baccatin III)[1]。编码DBAT 的基因最初由 Walker 和 Croteau[2]从东北红豆杉()悬浮细胞 cDNA 中克隆得到,并在大肠杆菌内实现了该酶的异源表达及功能鉴定。DBAT 是目前本实验室建立的一锅法双酶促反应从紫杉醇结构类似物 7-木糖-10-去乙酰紫杉醇(7-β-xylosyl-10-deacetytaxol,XDT)制备紫杉醇体系中的限速酶[3-5]。由 XDT 水解产生的 10-去乙酰紫杉醇(10-deacetytaxol,DT)与 10-DAB 只在 C13 位侧链上存在差异,但该差异导致 DBAT 水解 DT 的活性远低于其水解 10-DAB 的活性[3]。在前期研究中,我们推测并验证了 DBAT 中可能参与底物催化的一些重要位点,还获得了催化 DT 效率相比野生型提高近 6 倍的双突变体 DBATG38R/F301V,该双突变体被成功应用到一锅法反应,在 50 ml 反应体系中紫杉醇产量达到 0.64 mg/ml[3]。但相对于第一步反应中的糖基水解酶 LXYL-P1-2[6-7],DBATG38R/F301V的活性仍有待于进一步提高。前期研究结果发现,Ser396位于酶活性口袋内,但该位点可能不属于活性中心[3],可以用于定点突变研究以筛选活性得以提高的突变体。本文采用迭代饱和突变的方法,在 DBATG38R/F301V的基础上对该位点进行半饱和突变,以期发现活性比 DBATG38R/F301V进一步提高的突变株。同时,我们还对 DBAT 活性中心附近的剩余“热点”氨基酸残基进行了丙氨酸扫描研究,以期确定它们的功能,或发现活性得以提高的突变体。
1 材料与方法
1.1 材料
1.1.1 质粒和菌株 pCWori 表达载体由本室保存;pCWori-(pCWori 中连有东北红豆杉来源的,且 N 端连有 6 × His tag)和 pCWori-G38R/F301V(pCWori-第 38 位的 Gly 突变为 Arg,第 301 位 Phe 突变为 Val)由本实验室构建;大肠杆菌()Trans109 感受态细胞购自北京全式金生物技术有限公司。
1.1.2 试剂TransStart FastPfu FlyDNA polymerase 购自北京全式金生物技术有限公司;镍亲和层析填料购自美国 GE Healthcare 公司;其他试剂、溶液、酶以及培养基均见参考文献[3]。
1.1.3 仪器 Pro PCR 仪购自德国 Eppendorf 公司;mini transfer 购自美国 Bio-Rad 公司;Biospectrum AV system 购自美国 UVP 公司;3K15 型和 3K18 型低温高速离心机购自美国 Sigma 公司;P300 型超微量分光光度计购自德国 Implen 公司;全温振荡摇床购自哈尔滨东联科学仪器有限公司;FE20 型 pH 计购自瑞士 Mettler Toledo 公司;Labsolution 分析型高效液相(HPLC)分析工作站购自日本岛津公司;APV-2000 型高压细胞破碎仪购自德国 SPX 公司;HDL 超净工作台购自北京东联哈尔仪器制造有限公司;超纯水仪购自上海同田生物技术有限公司;Enspire 多标记微孔板检测仪购自美国 PerkinElmer 公司;电热恒温水箱购自北京市长风仪器仪表公司。
1.2 方法
1.2.1 丙氨酸扫描突变 参考文献[3]方法,构建相应位点的丙氨酸扫描突变体质粒,模板为pCWori-。PCR 引物序列如表 1 所示,突变后的密码子以下划线表示,“F”代表上游引物,“R”代表下游引物,所有引物均由北京睿博兴科生物技术有限公司合成。反应体系为50 μl,包括5 × TransStart FastPfu Fly buffer 10 μl,2.5 mmol/L dNTPs 5 μl,上下游引物各 2 μl,DNA 模板 5 ~30 ng,TransStart FastPfu Fly DNA 聚合酶 1 μl,加 ddH2O 至 50 μl。反应条件:95 ℃ 2 min,1 个循环;95 ℃ 10 s,55 ℃ 20 s,72 ℃ 2 min,共 30 个循环;72 ℃ 5 min。PCR 产物经 1% 琼脂糖凝胶电泳鉴定并纯化。纯化后的 PCR 产物在 37℃经I 酶切 3 h 后,转化大肠杆菌 Trans109 感受态细胞。
1.2.2 迭代饱和突变 即以pCWori-G38R/F301V为模板,以引物 S396X-F 和 S396X-R(表 1)进行全质粒PCR 扩增,在原来双突变的基础上引入396 位点的半饱和突变,构建一系列突变体质粒。PCR 反应条件同 1.2.1。

表 1 突变体及相应引物
1.2.3 突变体菌株培养和蛋白诱导表达 突变体菌株培养和蛋白诱导表达及蛋白纯化和浓度测定参照文献[3]。
1.2.4 巴卡亭III和紫杉醇的标准曲线绘制 精密称取巴卡亭 III 10.0 mg,色谱纯甲醇溶解,配制一系列浓度梯度:500、250、125、62.5、31.25 μmol/L;精密称取紫杉醇 5.30 mg,甲醇溶解,配制一系列浓度梯度:125、62.5、31.25、15.625、7.8125、3.90625 μmol/L。
HPLC 分析条件:色谱柱为Cosmosil-C18,流速为 1 ml/min,柱温为 28 ℃,检测波长为 230 nm,进样量 50 μl,流动相为乙腈和水,梯度洗脱条件如表 2 所示。根据峰面积和对应的浓度,得到以浓度为横坐标(X)、峰面积为纵坐标(Y)的回归方程。

表 2 梯度洗脱条件
巴卡亭 III 的线性回归方程为:Y = 82225X + 1E + 06,2= 0.9996;紫杉醇的的线性回归方程为:Y = 91206X – 11087,2= 0.9999。
1.2.5 突变体催化 DT 的最适条件测定 DBAT 突变体催化 DT 的最适 pH 和最适温度的测定反应体系同文献[3]。最适 pH 测定的反应条件为37.5 ℃;最适温度测定的反应条件为 pH 7.5(50 mmol/L 的磷酸钠缓冲液);均反应 3 h 后加入 400 μl 无水甲醇终止反应,HPLC 检测 DT 转化情况,HPLC 条件同 1.2.4。
1.2.6 突变体的热稳定性 将上述反应体系置于 50 mmol/L pH 7.5 的磷酸钠缓冲液中,分别于 37.5 ℃下静置 0、3、6 h,0 ℃下静置 0、2、4、6 h,按 1.2.5 所述方法测定酶催化 DT 的活性。
1.2.7 突变体催化 DT 的活性测定 突变体催化 DT 的活性测定参照文献[3]。
1.2.8 DBAT 突变体与底物 DT 的分子对接分析 在前期获得的野生型 DBAT 模拟三维结构和 docking 结果的基础上,利用 Chimera 软件中的 Tools → Structure editing → Rotamers 工具对野生型 DBAT 三维结构中的相关位点进行突变,突变后的氨基酸残基选择能量最优构象,分析突变后的氨基酸残基与底物 DT 的相互作用。
2 结果
2.1 Ser396半饱和突变体蛋白的活性检测
为节约筛选时间,提高筛选效率,我们通过迭代饱和突变(ISM)的方法[8]直接在DBATG38R/F301V的基础上继续进行Ser396位点的半饱和突变。经过测序验证,共获得了 10 个Ser396位点突变体,其中 S396R(为方便叙述,DBATG38R/F301V/S396R简称为 S396R,下同)、S396N、S396D、S396H、S396Y、S396G、S396C 和 S396V 没有检测到对非天然底物 DT 的催化活性,S396L 突变体依然保留着对非天然底物 DT 的催化活性,但活性有所下降,S396I 对DT 的催化活性有显著提高,分别达到 DBATG38R/F301V的 1.6 倍和野生型 DBAT 的 5.8 倍(图 1)。
2.2 DBATG38R/F301V/S396I催化 DT 的最适条件和热稳定性
对获得的高活性突变体蛋白进行进一步研究,如图 2A、B 所示,突变体蛋白DBATG38R/F301V/S396I催化非天然底物 DT 的最适温度为 37.5 ℃,最适 pH 为 7.5。热稳定性显示,在 37.5 ℃静置 3 h后,DT 的相对转化率为初始值的 75%,在 37.5 ℃静置 6 h 后催化 DT 的相对转化率为初始值的 30%(图 2C),在 0 ℃静置 6 h 后催化 DT 的相对转化率为初始值的 70% 左右(图 2D),说明虽然该突变体的活性与双突变对照相比有显著提高,但其在 0 ℃以上条件下不够稳定,需要继续通过蛋白质工程提高其热稳定性和(或)进一步提高其活性。
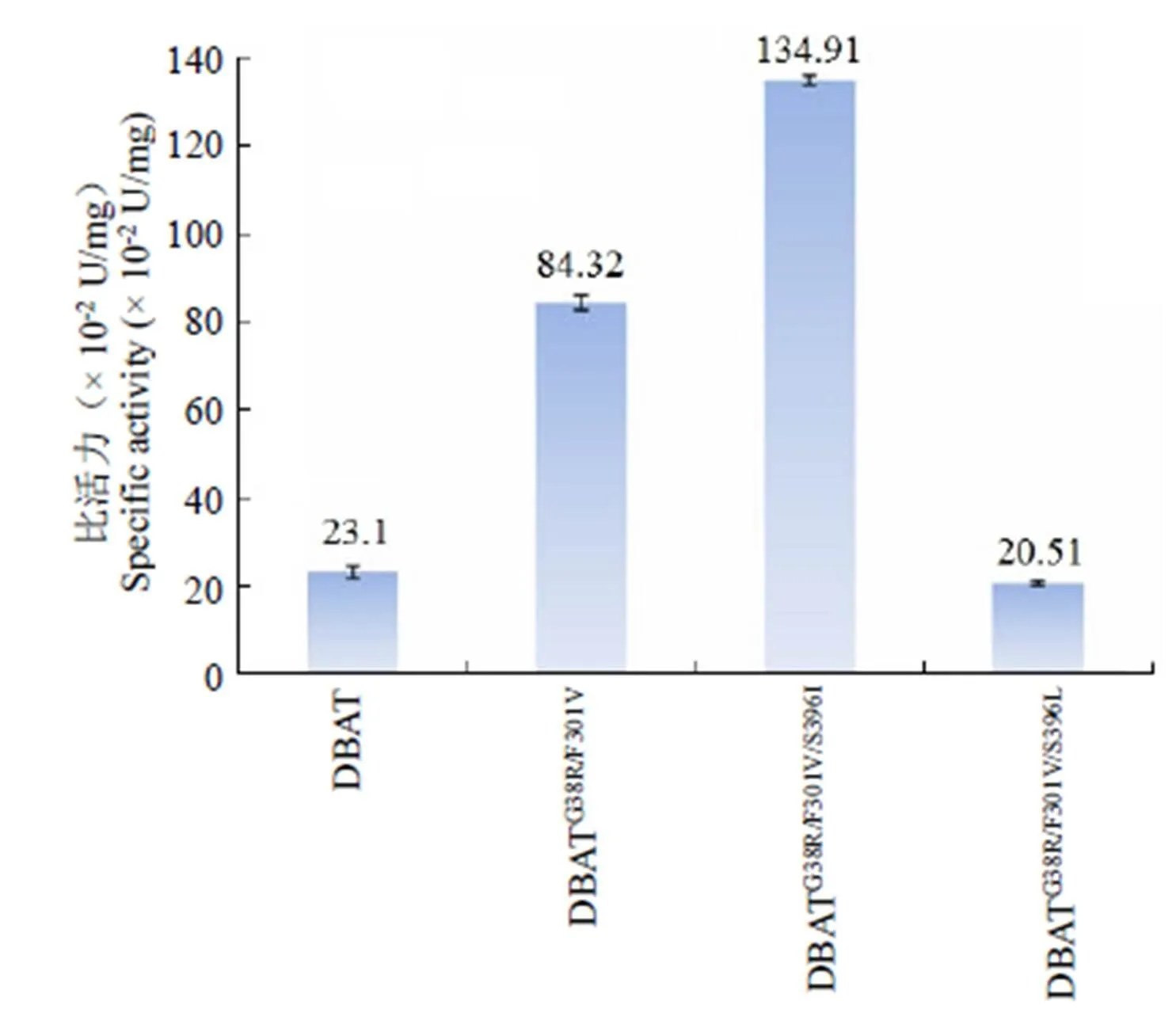
图 1 DBATG38R/F301V/S396I的活性检测
Figure 1 Activity of DBATG38R/F301V/S396I
2.3 结构分析
Ser396位于酶分子与底物结合口袋内部,但该位点不与底物直接相互作用(图 3A)。而 S396I 突变提高了酶对 DT 的催化活性,可能的原因是:Ser 为亲水性氨基酸,Ile 为脂肪族疏水性氨基酸,Ser396Ile 突变后增加了底物结合口袋的疏水性,该底物结合口袋和疏水性大分子底物 DT 的疏水性,相互作用增加,使底物更容易进入底物结合口袋,从而提高酶的催化活性。而 Ser396Leu 突变后活性却没有提高,推测氨基酸侧链的取向也对底物的催化有重要影响,Ile 的侧链取向(图 3B)可能更有助于底物的结合。
2.4 丙氨酸扫描突变
参考文献[3],在之前工作的基础上,我们选取了 DBAT 活性口袋附近的其他 5 个氨基酸位点进行丙氨酸扫描[9-10](图 4A)。将野生型 DBAT 的活性定义为 100%,各突变体蛋白催化天然底物 10-DAB 及非天然底物 DT 的活性测定结果如图 4B 所示。当以 10-DAB 为底物进行活性检测时,发现 N45A 位点突变造成活性完全丧失,N42A、E350A、E355A 和 K389A 突变的活性降低为对照的 20% 左右,说明除了本实验室之前已确定的一些氨基酸残基之外,Asn42、Asn45、Glu350、Glu355和 Lys389也可能参与了该酶对 10-DAB 的结合或催化。与上述结果相类似,当以 DT 为底物进行活性检测时,N45A 活性也完全丧失,N42A 和 E350A 突变活性下降约一半,E355A 和 K389A 突变体的活性下降较小。

图 2 DBATG38R/F301V/S396I 的最适反应条件和热稳定性(A:DBATG38R/F301V/S396I催化DT 的最适温度;B:DBATG38R/F301V/S396I催化DT 的最适 pH;C:DBATG38R/F301V/S396I在37.5 ℃的稳定性;D:DBATG38R/F301V/S396I在0 ℃的稳定性)
Figure 2 Optimal condition and thermal stability of DBATG38R/F301V/S396I(A: Optimal temperature of DBATG38R/F301V/S396Iagainst DT; B: Optimal pH of DBATG38R/F301V/S396Iagainst DT; C: Thermal stability of DBATG38R/F301V/S396Iat 37.5 ℃; D: Thermal stability of DBATG38R/F301V/S396Iat 0 ℃)

图 3 DBATG38R/F301V(A)和 DBATG38R/F301V/S396I突变体(B)与 DT 的分子对接局部图
Figure 3 Docking results of DBATG38R/F301V(A) and DBATG38R/F301V/S396I(B) with DT
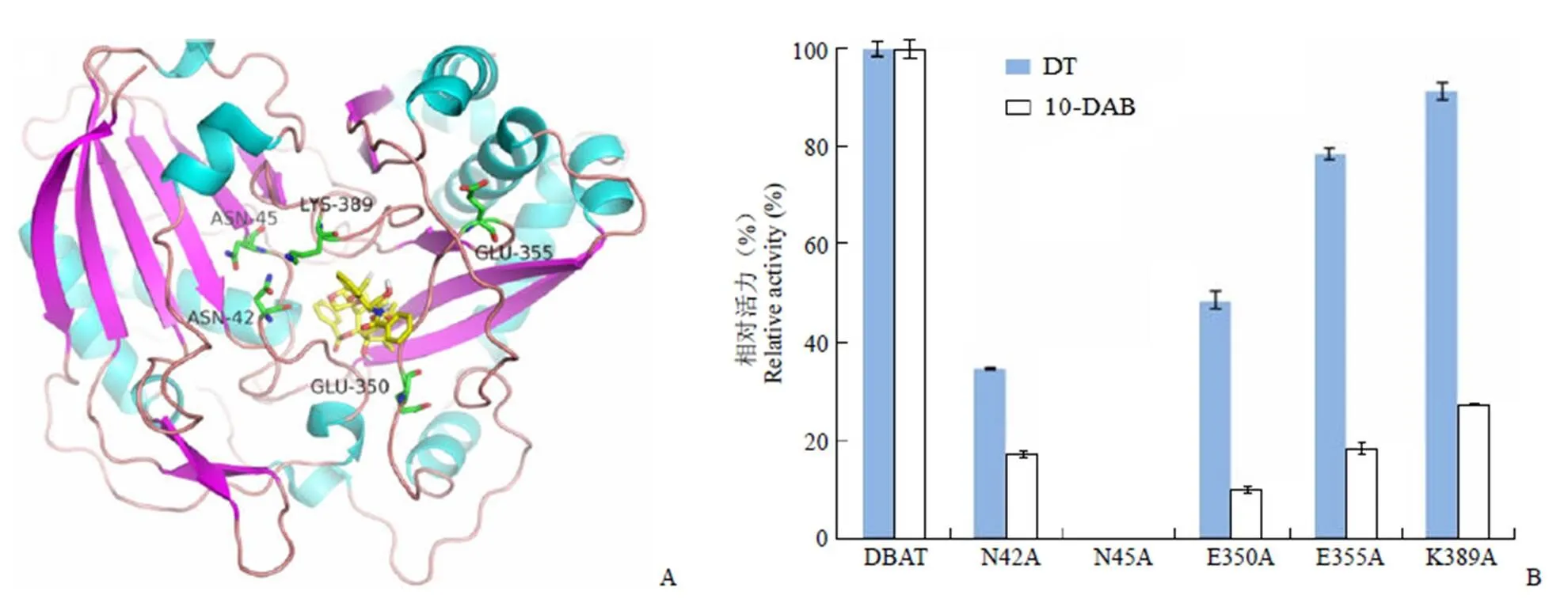
图 4 丙氨酸扫描突变分析(A:DBAT 三维结构分析;B:突变体活性分析)
Figure 4 Analysis of L-alanine scanning mutagenesis (A: Three-dimensional structure analysis of DBAT; B: Activity analysis of mutants)
3 讨论
半理性设计所需突变体的库容量远少于定向进化随机突变体的库容量,可大大减少筛选工作量,但通常仍需要借助高通量筛选的方法对突变体库进行筛选。文献显示,使用 NDT 简并密码子进行饱和突变时,单点突变的库容量仅为 34,远低于使用 NNK 兼并密码子时的库容量 94,且同时进行多点饱和突变时使用 NDT 简并密码子的优势尤为明显,能有效减少库容量,显著增加有益突变体出现的概率[8]。本文即采用 NDT 简并密码子进行(半)饱和突变研究。迭代饱和突变是在选定区域内进行反复饱和突变,在得到性能提高的突变体的基础上再次进行饱和突变,最终得到性能最优的突变体的方法[11-12],本研究中,使用该方法在已有的活性提高的双突变蛋白 DBATG38R/F301V的基础上,成功获得了活性进一步提高的三突变蛋白 DBATG38R/F301V/S396I。该突变体的最适温度为 37.5 ℃,与突变之前的 DBATG38R/F301V相同;但其最适 pH 为 7.5,高于 DBATG38R/F301V的最适 pH5.5。由于温度是进行酶促反应的一个重要因素,本研究重点观察了 DBATG38R/F301V/S396I的热稳定性,发现该蛋白对热不稳定,因此,还需要继续通过蛋白质工程提高其热稳定性和(或)进一步提高其活性。
通常位于酶活性口袋内部或其附近的氨基酸突变会直接影响酶的活性口袋的空间结构、电荷分布或疏水性,从而改变酶的催化特性。通过对 DBAT 活性口袋内部或其附近的一些氨基酸进行突变研究有可能发现对 DT 催化活性进一步提高的突变体。Ser396位于酶分子与底物结合口袋内部,在实验室前期的丙氨酸扫描结果中,其突变为丙氨酸之后活性约为野生型的 90%[3],说明该位点可能不参与活性中心的构成,所以在此基础上对其进行半饱和突变以期发现活性提高的突变株。DBATG38R/F301V/S396I突变体在保持其他氨基酸不变的前提下,将亲水性的 Ser 突变为疏水性的脂肪族氨基酸 Ile,结果表明,这种由亲水性向疏水性氨基酸的突变可能有利于该酶对 DT 的催化。但侧链的长度或侧链的空间构型也可能影响其活性,比如,同样是脂肪族氨基酸,当 396 位突变为 Leu 后,该酶的活性不升反降。又例如,该位点突变为 Val 和 Gly 后则活性完全丧失。
由于丙氨酸的 R-基团仅包含一个甲基,同时具有体积小、对蛋白质结构的影响较小的特点,因此“丙氨酸扫描技术”常被用于探究酶的活性位点[9-10],有时丙氨酸扫描还能发现活性位点之外使酶的催化活性得到提高的点突变[3]。活性口袋内丙氨酸扫描后活性降低的位点通常被认为是可能的底物结合(催化)位点。本研究中,我们通过丙氨酸扫描发现,Asn45位点对于两种底物的催化都至关重要,有可能是该酶的底物结合或催化位点之一,进一步完善了本课题组之前的研究结果。而其他位点的突变对于 10-DAB 和 DT 的影响并不完全相同,这一现象再次表明,虽然 DT 与 10-DAB 母核相同,但由于 DT 的 C13 位侧链的存在,造成其与 DBAT 结合后的空间取向与 10-DAB 存在差异,进而造成各个位点突变后突变体对两种底物的催化活性改变亦不尽相同。今后拟开展 DBAT 对 DT 的 C13位侧链适配性的研究,通过改变该酶活性腔的空间结构,使 DT 更容易进入该活性腔,筛选活性和(或)稳定性得到大幅度提高的 DBAT 突变体。
本研究通过改造酶活性口袋的催化环境,提高了其对 DT 的催化效率,加深了对 DBAT 突变体和 DT 相互作用机制的理解,为进一步开展 DBAT 的蛋白质工程研究奠定基础。
[1] Croteau R, Ketchum RE, Long RM, et al. Taxol biosynthesis and molecular genetics. Phytochem Rev, 2006, 5(1):75-97.
[2] Walker K, Croteau R. Molecular cloning of a 10-deacetylbaccatin III-10-O-acetyltransferase cDNA from Taxus and functional expression in Escherichia coli. Proc Natl Acad Sci U S A, 2000, 97(2): 583-587.
[3] Li BJ, Wang H, Gong T, et al. Improving 10-deacetylbaccatin III-10-β-O-acetyltransferase catalytic fitness for Taxol production. Nat Commun, 2017, 8:15544.
[4] Chattopadhyay SK, Sharma RP, Kumar S. Process for the production of important taxol analogues 10-deacetyl taxol A, B, and C: US, 6028206. 2000-02-22.
[5] Sisti NJ, Swindell CS, Chander MC. Paclitaxel synthesis from precursor compounds and methods of producing the same: US, 5948919. 1999-09-07.
[6] Cheng HL, Zhao RY, Chen TJ, et al. Cloning and characterization of the glycoside hydrolases that remove xylosyl groups from 7-β-xylosyl-10-deacetyltaxol and its analogues. Mol Cell Proteomics, 2013, 12(8):2236-2248.
[7] Zhu P, Cheng HL, Zhao RY, et al. Glycosyl hydrolase with beta-xylosidase and beta-glucosidase activities and uses thereof: US, 9206405. 2015-12-08.
[8] Reetz MT, Kahakeaw D, Lohmer R. Addressing the numbers problem in directed evolution. Chembiochem, 2008, 9(11):1797-1804.
[9] Cotter PD, Deegan LH, Lawton EM, et al. Complete alanine scanning of the two-component lantibiotic lacticin 3147: generating a blueprint for rational drug design. Mol Microbial, 2006, 62(3):735-747.
[10] Nicole P, Couvineau P, Jamin N, et al. Crucial role of the orexin-B C-terminus in the induction of OX1 receptor-mediated apoptosis: analysis by alanine scanning, molecular modelling and site-directed mutagenesis. Br J Pharmacol, 2015, 172(21):5211-5223.
[11] Reetz MT, Carballeira JD, Vogel A. Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability. Angew Chem Int Ed Engl, 2006, 45(46):7745-7751.
[12] Reetz MT, Bocola M, Carballeira JD, et al. Expanding the range of substrate acceptance of enzymes: combinatorial active-site saturation test. Angew Chem Int Ed Engl, 2005, 117(27):4264-4268.
Iterative saturation mutagenesis and active site analysis of 10-deacetylbaccatin III-10-β--acetyltransferase
ZHANG Yu-nan, CHEN Tian-jiao, ZHU Ping
State Key Laboratory of Bioactive Substance and Function of Natural Medicines & Key Laboratory of Biosynthesis of Natural Products of the National Health Commission, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, China
The study aims to improve the enzyme activity of DBATG38R/F301Vthrough iterative saturation mutagenesis, and identify the catalytic/substrate binding sites of DBAT by L-alanine scanning method.
Iterative saturation mutagenesis method was used to increase the DBAT activity against 10-deacetyltaxol (DT) through hemi-saturation mutation of Ser396on DBATG38R/F301V. The optimal reaction temperature and pH of the obtained mutant against DT were determined. Under the optimal pH its thermal stability was detected during the incubation period for up to 6 h at the optimal temperature and 0 ℃, respectively. The molecular mechanism for the improved enzyme activity was proposed based on the bioinformatics technology. L-alanine scanning mutagenesis was used to investigate the amino acids near the DBAT substrate binding region for their functions in catalyzing 10-DAB and DT.
A triple mutant DBATG38R/F301V/S396Iwith improved activity was obtained, which showed 1.6 times DT hydrolysis activity compared to the double mutant DBATG38R/F301V. It had an optimal temperature of 37.5 ℃ and an optimal pH of 7.5. In the thermal stability assay, it showed 30% of initial activity after incubation at 37.5 ℃ for 6 h, and maintained 70% of initial activity after incubation at 0 ℃ for 6 h. The Asn45of DBAT was determined as a crucial binding site for both 10-DAB and DT by L-alanine scanning of DBAT, while Asn42, Glu350, Glu355and Lys389were found to have more impact on the conversion of 10-DAB by DBAT.
Mutation from the hydrophilic amino acid to the hydrophobic aliphatic one in the active cave may help improve DBAT activity on DT. At the same time, the length and spatial configuration of the side chain of the amino acid are also important in affecting the enzyme activity; The Asn45was found to be a potential catalytic or substrate binding site of DBAT.
10-Deacetylbaccatin III-10-β--acetyltransferase; Iterative saturation mutagenesis; Highly active mutant; L-alanine scanning; Potential active site
ZHU Ping, Email: zhuping@imm.ac.cn
国家自然科学基金(81573325、31270796);北京协和医学院“协和青年科研基金”(3332015137);中国医学科学院医学与健康科技创新工程重大协同创新项目(CIFMS-2017-I2M-2-004);中央高校基本科研业务费专项资金(2017PT35001)
朱平,Email:zhuping@imm.ac.cn
2018-08-30
10.3969/j.issn.1673-713X.2018.06.001
