柱前衍生-超高效液相色谱-串联四极杆质谱法测定茶叶中乙撑硫脲的残留
2018-12-06郑小严
郑小严
(福建省产品质量检验研究院, 福建 福州 350002)
乙撑硫脲(ethylenethiourea,简称ETU)是代森类杀菌剂(ethylenebisdithioearbomates,简称EBDCs)的共同降解产物及其产品的杂质。ETU具有慢性毒性,对哺乳动物具有致癌性、致突变性及致畸性。虽然ETU在农作物和农产品中的残留量相对较低,但不少研究表明,残留在农产品中的EBDCs可在农产品的加工、烹饪等过程中转化为ETU,从而带来危害[1]。代森锰锌因其具有高效、广谱等优点而在农作物病虫防治过程中广为应用,如在茶叶种植中用于防治赤星病[2],但有关研究[3]显示,长期暴露于代森锰锌可能与帕金森等精神退化性疾病相关。《食品安全国家标准中食品农药的最大残留量》[4]中代森锰锌的每日最大允许摄入量(ADI)为0.03 mg/kg BW,但对乙撑硫脲的残留限量还没有规定;叶孟亮等[5]研究了我国苹果中乙撑硫脲的残留水平并进行了膳食摄入风险评估,最后建议将中国苹果中乙撑硫脲最大残留限量值设为0.2 mg/kg。因此对食物中代森锰锌及其代谢物乙撑硫脲的检测显得很有必要。
关于乙撑硫脲的残留分析方法已有很多研究报道。目前,ETU的相关检测方法主要有GC、GC-MS、HPLC和HPLC-MS/MS法等多种。但由于ETU具有很强的极性,很难用GC直接测定其残留量,通常是先将其用氯化苄转化为低沸点的S-苄基ETU,再用带氮磷检测器(NPD)的气相色谱仪[6-8]或气相色谱-质谱联用仪[9]进行测定,其操作程序非常复杂,衍生不完全带来的误差大。采用液相色谱法检测[10-14]时,由于极性大、出峰快,经常存在较大的干扰,需要复杂的净化方法[10],并且紫外检测器的灵敏度也不够理想。此外,采用液相色谱-质谱联用法[15,16]检测时,茶叶等复杂样品基体存在严重的离子抑制作用,大大降低了检测的灵敏度;Lindh等[17]采用五氟苄基溴柱前衍生-液相色谱-质谱联用法检测人尿液中的ETU残留,该方法大大提高了检测的灵敏度,但其衍生及提取操作较复杂。本研究针对现有乙撑硫脲检测方法的缺点,采用9-芴基甲基氯甲酸酯(FMOC-CL)为衍生剂,对衍生条件、提取方法和检测条件等进行了深入的探讨与研究,建立了柱前衍生-超高效液相色谱-四极杆质谱(UPLC-MS/MS)检测茶叶中乙撑硫脲残留的方法。该方法简便、快捷、灵敏,可以更高效地满足茶叶中乙撑硫脲的检测需求。
1 实验部分
1.1 仪器与试剂
超高效液相色谱Waters Acquity UPLC系统配Quattro Premier XE四极杆质谱仪(美国沃特世公司);冷冻高速离心机(Avanti J-E,美国贝克曼公司);涡旋混合仪(美国热电公司);分析天平(BSA224S,赛多利斯北京有限公司); Milli-Q超纯水纯化系统(美国Millipore公司);超声波清洗器(KQ-800KDE,昆山市超声仪器有限公司);聚四氟乙烯(PTFE)一次性过滤头(0.2 μm,美国热电公司)。
乙撑硫脲标准品(纯度98.5%,德国Dr. Ehrenstorfer公司);乙撑硫脲-D4同位素内标(纯度99.3%,加拿大CDN Isotopes公司); 9-芴基甲基氯甲酸酯(色谱纯,纯度≥ 97.0%,上海安谱实验科技股份有限公司);乙腈(色谱纯,山东禹王实业有限公司化工分公司);石墨化碳黑(GCB,美国安捷伦公司);无水硫酸镁(分析纯,国药集团化学试剂有限公司);实验用水均为Milli-Q超纯水纯化系统制备的超纯水。
1.2 试剂的配制
衍生剂配制:称取1.0 g FMOC-CL,用乙腈溶解,定容到10 mL,配成100 g/L溶液。
QuEChERS基质分散固相萃取净化管:称取50 mg GCB和500 mg无水硫酸镁于2 mL子弹头离心管中备用。
乙撑硫脲及乙撑硫脲-D4内标标准储备溶液的配制:分别准确称取适量乙撑硫脲及乙撑硫脲-D4标准品,用乙腈溶解配制成约500 mg/L的标准储备溶液。
乙撑硫脲及乙撑硫脲-D4内标标准中间溶液的配制:分别准确移取代乙撑硫脲及乙撑硫脲-D4标准储备溶液1.00 mL,用乙腈定容到50 mL,配制成10.0 mg/L的标准中间溶液。
乙撑硫脲标准使用溶液的配制:准确移取适量乙撑硫脲标准中间溶液和内标溶液,配成乙撑硫脲质量浓度为1~200 μg/L、乙撑硫脲-D4质量浓度均为100 μg/L的系列标准溶液。
1.3 样品处理方法
准确称取1.00 g粉碎均匀的茶叶样品,加入10.0 mL乙腈和100 μL 10.0 mg/L的乙撑硫脲-D4内标溶液,振摇混匀,超声提取30 min,摇匀,4 ℃、10 000 r/min下高速离心3 min;吸取1.5 mL上清液注入QuEChERS净化管中,振摇1 min混匀,16 000 r/min下高速离心3 min;准确移取上清液1.0 mL于5 mL刻度试管中,加入200 μL 100 g/L FMOC-CL乙腈溶液,立刻涡旋混匀,避光静置60 min,过0.2 μm PTFE滤膜,6 h内检测。
1.4 色谱和质谱条件
1.4.1超高效液相色谱条件
色谱柱:Waters BEH C18(100 mm×2.1 mm, 1.7 μm);柱温:35 ℃;进样量:10 μL;流速:0.3 mL/min;流动相:A为0.1%(v/v)甲酸溶液,B为乙腈。洗脱梯度:0~6.0 min, 40%A~20%A; 6.0~6.1 min, 20%A~5%A; 6.1~8.0 min, 5%A; 8.0~8.1 min, 5%A~40%A; 8.1~10.0 min, 40%A。
1.4.2质谱条件
电离源:大气压电喷雾离子源正离子模式;毛细管电压:3.50 kV;源温度:120 ℃;脱溶剂气温度:400 ℃;脱溶剂气流量:700 L/h;锥孔反吹气流量:50 L/h;碰撞室压力:0.27 Pa;电子倍增管电压:650 V;检测方式:多反应监测(MRM)模式;特征离子等参数见表1。

表 1 乙撑硫脲及其同位素内标的9-芴基甲基氯甲酸酯衍生物的质谱参数
Parent ion: [M+H]+; * quantitative ion.
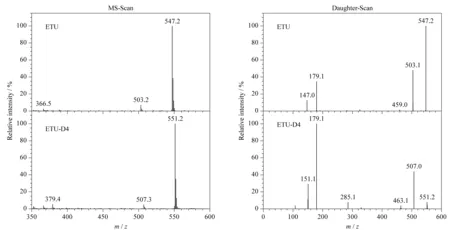
图 1 乙撑硫脲衍生物和乙撑硫脲-D4衍生物的质谱图
2 结果与讨论
2.1 质谱条件的优化
取约1 mg/L的乙撑硫脲标准溶液1.0 mL,加入200 μL衍生剂衍生。将衍生后的标准溶液由进样器进样,经色谱柱分离后采用MS-Scan模式进行一级质谱扫描,在ESI(+)模式下找到乙撑硫脲二取代衍生物准分子离子峰m/z547.2;再经母离子碰撞后采用Daughter-Scan模式进行二级质谱子离子扫描,通过优化碰撞能量等参数得到乙撑硫脲衍生物的子离子扫描质谱图,同法对乙撑硫脲-D4同位素内标的衍生物进行一级和二级质谱扫描,结果的质谱图见图1,最终确定相应的质谱参数,见1.4.2节。
2.2 色谱条件的优化
本方法采用了美国Waters公司的超高效液相色谱仪和1.7 μm粒径的BEH-C18超高效液相色谱柱,具有极高的分离效率。本研究分别考察了纯水/乙腈体系、0.02%(v/v)甲酸/乙腈体系、0.1%(v/v)甲酸/乙腈体系、0.3%(v/v)甲酸/乙腈体系作为流动相的分离效果,发现提高流动相酸度有利于提高离子化效率和检测灵敏度,但酸度太大会发生色谱峰开叉,最终发现在0.1%(v/v)甲酸/乙腈体系下检测效果最佳。优化后的色谱条件和洗脱梯度见1.4.1节。此时,乙撑硫脲及同位素内标标准溶液衍生后的总离子流(TIC)图和MRM色谱图见图2。
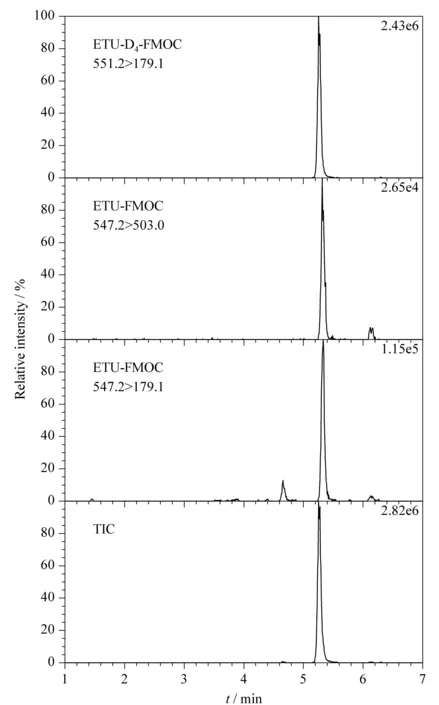
图 2 乙撑硫脲(5 μg/L)及ETU-D4(100 μg/L)衍生物的总离子流图和MRM色谱图
2.3 衍生化反应条件的优化
9-芴基甲基氯甲酸酯以其反应产物稳定、反应速度快且完全而成为一种十分理想的氨基衍生试剂,常用于氨基酸的衍生测试。实验发现,乙撑硫脲的两个氨基上的氢均会被FMOC-CL基团取代,衍生产物相对分子质量的理论值为546.0,与质谱扫描发现的m/z547.2分子离子峰完全吻合,同时,碰撞后产生m/z503.0、459.0和179.1的碎片离子,其反应路径见图3。由于茶叶中存在大量的茶氨酸等氨基酸类物质,这些氨基酸均会与FMOC-CL发生衍生反应,为保证衍生剂的量充足,本实验在1 mL提取液中加入200 μL质量浓度为100 g/L FMOC-CL乙腈溶液进行衍生反应。文献[18]中FMOC-CL的衍生条件通常是加入硼砂缓冲液,在偏碱性条件下反应,过量的衍生剂会与水反应生成FMOC-OH和二酰化产物。实验中发现在含水较多的条件下衍生时,乙撑硫脲的衍生产物峰面积较小并且稳定性很差,半个小时内就会发生50%的衰减。而在无水的条件下,即无需加入硼砂缓冲液,乙撑硫脲也能与FMOC-CL发生衍生反应,衍生产物的峰面积明显大于加入缓冲液时衍生产物的峰面积(见图4),并且衍生产物稳定性大大提升(见图5),衍生反应在1 h左右达到平衡,6 h内没有明显变化,但12 h后降解为最大值的60%左右。因此,本方法中茶叶的乙腈提取液不加缓冲液,直接衍生,并且在样品衍生1 h后和6 h内进行样品测试。造成该现象的原因可能是,乙撑硫脲与FMOC-CL的反应速率远低于FMOC-CL与水反应的速率,FMOC-CL大量与水反应造成乙撑硫脲衍生不完全,衍生产物在大量水分子的存在下会极不稳定,降解速度很快,为了维持衍生反应体系的稳定性,应尽可能减少水分子的存在。由于乙撑硫脲衍生产物的质量浓度会随时间发生变化,因此,该方法必须采用同位素内标法进行校正。
2.4 样品提取条件的选择
乙撑硫脲的极性较大,水是最佳溶剂。目前,文献采用的提取剂主要为甲醇-水溶液[6,7,11,14]和氨水-乙腈溶液[10,13]等,这些提取剂中都含有水。由于本研究发现,乙撑硫脲与FMOC-CL的衍生反应要尽可能避免水的存在,因此采用乙腈为提取剂。乙腈对乙撑硫脲也具有良好的溶解性,同时采用超声振荡提取的方法可加快样品的提取速度。表2的加标回收试验数据表明,该提取方法效果良好。

图 3 乙撑硫脲与FMOC-CL的衍生反应及其产物的碰撞碎裂路径

图 4 两种衍生方法下茶叶加标提取液的质谱结果
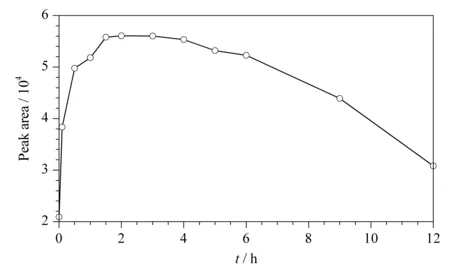
图 5 茶叶加标衍生产物峰面积的稳定性
2.5 样品净化条件的选择
茶叶样品水分含量较低,但含有大量的色素,未经净化的样品会造成较严重的离子抑制和色谱柱污染。为了去除茶叶乙腈提取液中的少量水分和大量色素,本实验采用简单易行的QuEChERS基质分散萃取净化方法,使用GCB吸附色素和无水硫酸镁吸附水分的组合方式净化样品提取液。比较10、20、50、100 mg GCB加200、500 mg无水硫酸镁的配方分别净化1.5 mL红茶、绿茶、普洱茶及铁观音茶的样品提取液。实验发现50 mg GCB已能完全吸附4种茶提取液中的色素,所得溶液澄清透亮;由于茶叶中水分普遍较低,200 mg和500 mg无水硫酸镁吸附水分后的提取液衍生效果没有明显区别,由于无水硫酸镁价格低廉,同时为了保证吸附水分的效果,本实验选择使用500 mg无水硫酸镁。最后确定在2 mL离心管中称取50 mg GCB和500 mg无水硫酸镁,加入1.5 mL茶叶提取液振摇净化,高速离心后取1.0 mL上清液衍生后测试。经净化后样品提取液色素去除明显,衍生产物稳定性达到6 h以上,4种茶叶加标提取液的色谱
峰响应值分别提高了2~4倍,该净化方法操作简便,适合大批量样品的检测。
2.6 方法的线性范围和检出限
在1.0~203.4 μg/L内,以进行衍生反应的乙撑硫脲标准工作溶液质量浓度为横坐标(X),以乙撑硫脲定量离子峰面积除以内标峰面积的值与内标质量浓度的乘积为纵坐标(Y),绘制标准工作曲线,所得线性方程的相关系数r为0.999 3。因此,在该浓度范围内线性关系良好。同时,用乙撑硫脲含量为10 μg/kg的茶叶加标样品峰高按3倍信噪比和10倍信噪比计算乙撑硫脲的方法检出限为1.3 μg/kg和定量限为4.2 μg/kg。
2.7 回收率和精密度试验
茶叶分发酵茶(如红茶和黑茶)、半发酵茶(如青茶,代表为铁观音)和不发酵或轻发酵茶(如绿茶、白茶和黄茶)。分别取不含乙撑硫脲的空白红茶、绿茶和铁观音茶样品进行标准添加试验,3个水平各做6个平行试验,结果见表2。该方法对乙撑硫脲的回收率在97.7%~107.5%之间,相对标准偏差在2.1%~10.0%之间。

表 2 茶叶加标样的回收率和精密度(n=6)
2.8 茶叶实际样品的检测
采用本方法对市售的13个绿茶样品、13个红茶样品和10个铁观音茶样品进行检测,36个茶叶样品均未检出乙撑硫脲残留。
3 结论
本文建立了茶叶中的乙撑硫脲经FMOC-CL柱前衍生化反应后利用超高效液相色谱-串联四极杆质谱检测的方法。所建立的分析方法前处理步骤简单,具有良好的线性关系和灵敏度,重现性好,定性定量准确,可有效满足茶叶中乙撑硫脲残留检测的要求,为推动相关国家标准的制定提供了可靠的技术支撑。
猜你喜欢
杂志排行

色谱的其它文章
- 获2017年度领跑者5000
--中国精品科技期刊顶尖学术论文入选证书的《色谱》论文名单 - Determination of airborne formaldehyde and ten other carbonyl pollutants using programmed temperature vaporization-large volume injection-gas chromatography
- 重组含糖识别结构域的人源半乳糖凝集素-3在糖蛋白/糖肽富集中的应用
- QuEChERS-液相色谱-串联质谱法测定蔬菜中105种农药残留
- 超高效液相色谱-串联质谱法测定热带水果中杀虫双残留
- 高效液相色谱-串联质谱法分析毛发中甲基苯丙胺和苯丙胺手性对映异构体