高丹草杂种及其亲本转录组SNP及等位基因特异性表达分析
2018-11-29逯晓萍张坤明薛春雷张瑞霞
董 婧 逯晓萍,* 张坤明 薛春雷 张瑞霞
高丹草杂种及其亲本转录组SNP及等位基因特异性表达分析
董 婧1逯晓萍1,*张坤明1薛春雷1张瑞霞2
1内蒙古农业大学农学院, 内蒙古呼和浩特 010019;2呼和浩特市种子管理站, 内蒙古呼和浩特 010020
为探究高丹草杂种及其亲本间单核苷酸变异与其杂种优势形成的关系, 以高丹草杂种及其亲本的根、茎和叶组织为试验材料, 采用Illumina Hiseq 2000进行转录组测序。对平均长度达58 122 160 bp的各测序样品序列信息进行检测后, 均检测到不少于58 000个SNP位点, 位于基因内的SNP个数显著多于位于基因间的SNP个数, SNP发生频率为1/741 bp, 平均转换颠换比为1.00∶1.53。在所有变异类型中, C/T和G/A发生频率最高。经过筛选得到198 (21%)个极显著偏向性等位基因表达偏向性SNP, 其中65%偏向父本白壳苏丹草, 并且很多在白壳苏丹草中具有高水平基因表达的转录本, 在高丹草杂种中的等位基因表达也偏向白壳苏丹草。在3种组织中, 分别有79%、78%和82%转录本的2个亲本等位基因表现出相对平衡的表达水平, 说明与顺式作用相比, 反式作用可能更多地影响了等位基因的特异性表达。选择6个极显著偏向性等位基因表达偏向性SNP-unigene进行qRT-PCR验证, 这些基因的差异基因表达模式与RNA-Seq分析结果一致。本研究采用Illumina 测序技术研究等位基因表达, 为高丹草杂种优势分析提供了依据, 也为其他饲草作物的相关研究提供了理论参考。
高丹草; 杂种优势; 转录组; 单核苷酸多态性; 功能注释
高粱()是一种古老的禾谷类作物, 具有抗旱、耐涝、耐盐碱、适应性强等特点, 苏丹草()是栽培最普遍的一年生禾本科牧草, 具有高度的适应性、很强的再生性和抗旱能力, 其茎叶品质优良[1]。高丹草是高粱-苏丹草杂交种的简称, 它结合了高粱和苏丹草的优点, 在畜牧业和渔业生产上具有广阔的开发利用前景[2]。这种优良的农艺性状表现可能是由2个亲本基因组互作造成的, 但是具体的遗传机制尚不清楚。
杂交是自然界中普遍存在的现象, 不仅可以对物种形成、适应性进化和生态创新产生重要作用, 而且可能出现大量的等位基因变异[3-7]。有研究表明, 融合这些等位基因变异可能导致新的基因行为方式的出现, 从而产生杂种优势[8-11]。但是, 以往对于杂种优势机理的研究只针对杂种及亲本基因的表达水平, 而对杂种中不同亲本等位基因差异表达的研究较少。
SNP (single nucleotide polymorphisms)是指在基因组上由单个核苷酸变异形成的遗传标记, 其数量庞大[12]。通常杂交种中的等位基因特异性表达的研究方法有2种[13], 一是基于标记多态性, 利用已知基因组变异获得高质量的等位基因表达结果[14-15]; 另外一种是利用SNP芯片同时获得上万个等位基因特异性表达位点[16-17]。然而, 这两种方法都必须提前知道研究对象的基因组信息。随着第二代高通量测序技术的发展, RNA-Seq技术逐渐被人们所熟悉并应用到等位基因特异性表达的研究中[18]。RNA-Seq技术无须预知研究对象的基因组信息并且分辨率能够达到单碱基水平。应用这个方法能够从全基因组水平无偏估计基因的调控, 而且能同时获得转录丰度和等位基因表达偏向性的信息[19]。
本研究在高丹草遗传图谱构建、产量性状QTL定位、杂种表现遗传模型以及差异表达基因分析与蛋白质组学等[2,20-23]研究的基础上, 比较和分析高丹草杂种中2个亲本等位基因的特异性表达, 旨在了解杂交种中双亲等位基因的不同作用以及可能对杂种优势的贡献, 进一步阐明高丹草杂种优势的分子机制, 为高丹草杂种的遗传改良提供依据。
1 材料与方法
1.1 植株材料及样品采集
以高丹草杂种(11A×白壳苏丹草)一代、母本高粱11A和父本白壳苏丹草的根、茎、叶为试材, 设置3个生物学重复(表1)。在三叶期, 分别取高丹草杂种及其亲本的根、茎、叶样品, 立即投入液氮冷冻, 于-80℃保存备用。
1.2 mRNA高通量测序及SNP分析
利用TRIzol法提取高丹草杂种及其亲本各组织的总RNA, 构建高质量文库后, 使用Illumina Hiseq 2000测序仪测序。由于高丹草和苏丹草均尚未完成基因组测序, 所以使用亲本之中已完成基因组测序的高粱基因组(ftp://ftp.jgi-psf.org/pub/compgen/phyt ozome/v9.0/early_release/Sbicolor_v2.1/)作为参考进行后续分析。利用TopHat 2[24]软件在Clean Reads和高粱参考基因组之间比对。

表1 材料编号及名称
在各样品读长与参考基因组序列的TopHat2软件比对数据的基础上, 利用GATK软件寻找测序样品与参考基因组间的单碱基错配[25]。根据Nr注释信息, 使用Blast 2 GO软件[26]和WEGO (Web Gene Ontology Annotation Plot)软件[27]得到unigene的GO (Gene Ontology)注释信息和功能分类统计。利用蛋白数据库KEGG (http://www.genome.jp/kegg/)进一步得到unigene的pathway注释[28]。通过blastx (e-value<10–5)将包含SNP位点的unigene比对到COG数据库, 从而获得其COG分类注释[29]。
本研究通过比较高丹草杂种及其亲本3种组织所有同源基因测序读长, 按照以下标准筛选单核苷酸多态性位点: (1)所有SNP位点须在高丹草及其亲本3种组织测序的3个重复中均出现; (2)为确保等位基因特异性表达数据的可靠程度, 各SNP位点至少有300条reads的支持。在某SNP位点, 两亲本reads中的碱基互不相同, 子代与亲本之一碱基相同, 则认为高丹草杂种中存在等位基因表达偏向性。符合上述标准的SNPs用于后续分析。如果杂交种中2个亲本等位基因对应的reads支持数的比值偏离1.0, 则认为杂交种中存在等位基因表达偏向性。采用卡方检验方法统计分析。
1.3 实时荧光定量PCR (Quantitative real-time PCR, qRT-PCR)
从极显著偏向性等位基因表达偏向性SNP- unigene中选择6个基因进行实时荧光定量PCR验证分析, 使用 E.Z.N.A.Plant RNA Maxi Kit抽提RNA。根据基因序列, 使用Primer Quest Tool (http:// sg.idtdna.com/ Primerquest/Home/Index)设计引物。第1链反转录使用PrimeScrip RT reagent Kit with gDNA Eraser (Perfect Real Time)第1链合成试剂(RR047A)。荧光定量检测使用abmEva Green qPCR Master Mix-No Dye试剂盒。PCR反应流程为95℃预变性1 min; 95℃变性10 s, 60℃退火30 s, 40个循环。以b-actin为内参, 使用2–ΔΔCt法分析表达量。
2 结果与分析
2.1 SNP位点统计
根据碱基替换的不同方式, 可以将SNP位点分为转换(Transition)和颠换(Transversion) 2种类型; 根据SNP位点的等位(Allele)数目, 可以将位点分为纯合型(只有一个等位)和杂合型(两个或多个等位)[30]。计算高丹草杂种及其亲本中的SNP位点数目、转换类型、颠换类型比例和杂合型SNP位点比例, 统计结果见表2。

表2 SNP位点统计表
(续表2)

材料编号Material number总读长Total readsSNP数SNP number基因内SNPGenic SNP基因间SNPIntergenic SNP转换Transition (%)颠换Transversion (%)杂合型Heterozygosity (%) 1759 733 16095 72389 741598260.1739.8333.54 1872 158 58081 26775 641562660.3439.6629.79 1978 016 50895 20587 506769960.5039.5036.17 2053 568 21882 07977 412466760.4739.5330.25 2149 779 34273 90569 659424660.7539.2530.17 2259 151 35689 06882 884618460.3639.6438.21 2350 348 06675 27670 970430660.2339.7736.31 2451 025 42472 97868 695428360.4839.5236.28 2555 095 66881 94576 448549760.7539.2531.16 2657 676 61287 67382 696497760.3739.6331.16 2761 078 42478 86474 174469060.7439.2630.43
材料名称及编号同上表1。
Name and number of the material are the same as there given in Table 1.
对平均长度达58 122 160 bp的序列, 各测序样品中均检测到不少于58 000个SNP位点, 位于基因内的SNP个数显著多于位于基因间的SNP个数。高丹草杂种及其亲本转录组SNP发生频率为1/741 bp, 即平均每741 bp就有1个SNP位点出现。另外, 各样品中转换类型的SNP均占所有SNP数目的60%以上, 明显多于颠换类型的SNP个数, 平均转换颠换比为1.00︰1.53 (表2)。
2.2 SNP类型
由图1可知, 在所有12种单核苷酸变异类型中, 发生频率最高的前4种分别是C/T、G/A、A/G和T/C, 均大于40 000个, 而其他8种单核苷酸变异C/G、G/C、C/A、G/T、T/G、A/C、A/T和T/A均在20 000以下。12种变异类型中以C/T类型频率最高, 原因可能是CpG二核苷酸上甲基化的胞嘧啶残基易脱去氨基而转化成胸腺嘧啶[31]。

图1 SNP类型统计
2.3 SNP注释
将SNP-unigene序列与GenBank中的非冗余蛋白数据库Nr数据库进行相似性比对。其中, 比对效率最高的有24 515条unigene (79.12%)与数据库中已知的高粱基因同源; 3806条unigene (12.28%)与数据库中已知的玉米基因组同源; 1435条unigene (4.63%)与数据库中已知的谷子基因组同源(图2)。
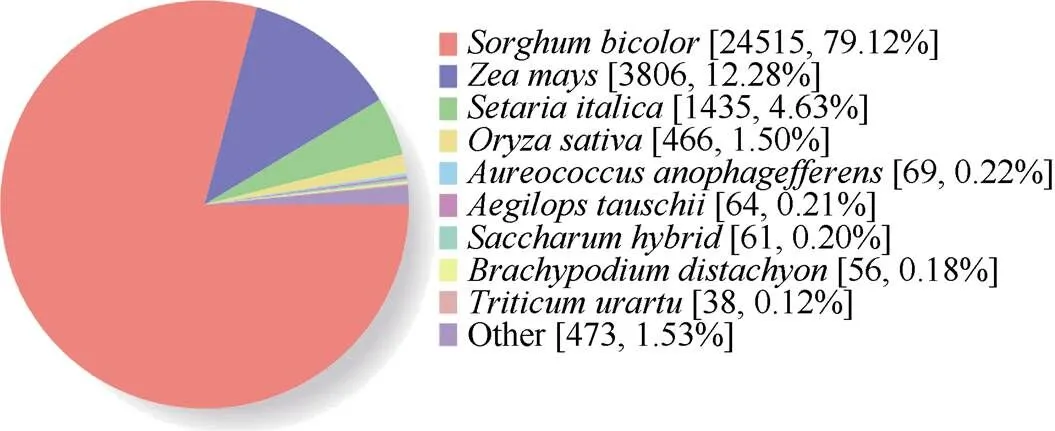
图2 SNP-unigene Nr比对结果
括号内为该类型unigene数及其在所有unigene中所占比例。
Unigene number of this type and its proportion in all unigene are shown in parentheses.
能够在COG中找到10 328条unigene相应的注释信息, 共获得15 799个COG功能注释, 可分为24类(图3中A~Z表示), 并对其进行数量统计。从分析统计结果可以看出, 这10 328条被注释的unigene功能种类较为全面, 涉及大多数生命活动过程或功能。“一般功能预测类”是最大的一个分类, 包含3099 (19.05%)个unigene。其次是“转录”、“复制、重组和修复”、“信号转导机制”和“翻译、核糖体结构和生物转化”分别包含1521 (9.63%)、1430 (9.05%)、1309 (8.29%)和1099 (6.96%)条SNP-unigene。“核结构”分类中包含2 (0.01%)条SNP-unigene, 数量最少(图3)。

图3 SNP-unigene COG比对结果
2.4 等位基因偏向性表达与亲本差异相关
为了分析杂种中的等位基因特异性表达, 比较得到注释的转录本外显子区域的每个碱基并鉴定SNP, 经过筛选后, 将9308个SNP用于后续分析。在本研究中, 如果在一个转录本中同时存在多个SNP, 并且其中2个SNP的表达偏向性不同, 则将该转录本信息直接删除。为了保证分析的准确性和可靠性, 本试验只将表现极显著偏向性(<0.01)的SNP用于后续分析。图4所示是198个极显著偏向性等位基因表达偏向性SNP在3种不同组织中的表达情况。
在亲本基因差异表达对高丹草杂种中的等位基因特异性表达方式的影响分析中, 将父本白壳苏丹草与母本高粱11A的基因表达比值命名为P1/P2, 高丹草杂种中的亲本等位基因表达比值命名为F1/P2。结果表明, 很多在白壳苏丹草中具有高水平基因表达的转录本, 在高丹草杂种中的等位基因表达也偏向白壳苏丹草(图5)。
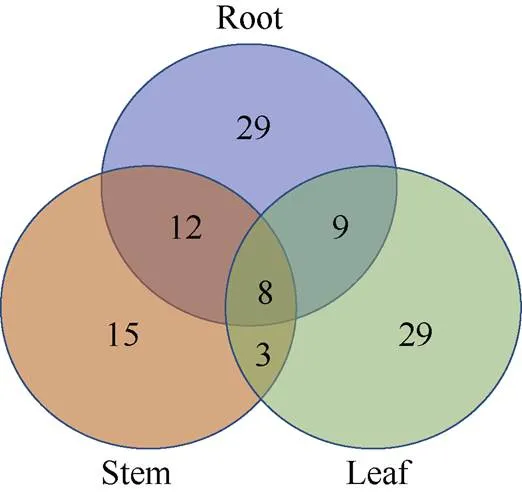
图4 等位基因表达偏向性SNP
在198个SNPs中, 根、茎、叶组织中分别有79个(涉及58个基因)、53个(涉及38个基因)和66个(涉及49个基因)单核苷酸多态性位点具有等位基因表达偏向性。其中, 有10个SNPs (涉及8个转录本)在根、茎、叶中均有表现(表3)。

图5 高丹草杂种中等位基因表达偏向性

表3 10个等位基因表达偏向性SNPs
GO功能分析将Sobic.001G191200转录本定位到蔗糖响应机制(response to sucrose)、葡萄糖响应机制(response to glucose)、果糖响应机制(response to fructose)功能; Sobic.001G293800转录本定位到蛋白质磷酸化(protein phosphorylation)、ATP结合(ATP binding)、叶绿体(chloroplast)等32种功能; Sobic.002G215700转录本定位到干旱响应机制(response to desiccation)、醛脱氢酶活性(aldehyde dehydrogenase, NAD)、胞液(cytosol)等17种功能; Sobic.003G085700转录本定位到RNA加工(RNA processing)、基因表达调控 (regulation of gene expression)和核腔(nuclear lumen)等7种功能; Sobic.003G206800转录本定位到脂肪酸β氧化(fatty acid beta-oxidation )、激酶活性(kinase activity)等12种功能; Sobic.003G314500转录本定位到水解酶活性(hydrolase activity)、叶绿体被膜(chloroplast envelope )、有机物质代谢过程(organic substance metabolic process )等5种功能; Sobic.004G225100转录本定位到叶绿体(chloroplast)、吲哚乙酸生物合成过程(indoleacetic acid biosynthetic process )、氰化物代谢过程(cyanide metabolic process)等20种功能; Sobic.004G253000转录本定位到胞膜界小泡(cytoplasmic membrane-bounded vesicle)功能。
KEGG代谢通量分析发现Sobic.001G293800转录本参与b录淀粉酶代谢通路; Sobic.002G215700转录本参与醛脱氢酶家族7成员A1代谢通路; Sobic. 004G225100转录本参与腈水解酶代谢通路; Sobic. 004G253000转录本参与FAM32A (A)蛋白代谢。
2.5 荧光定量PCR验证结果
从198个等位基因表达偏向性SNPs中随机选择6个基因进行qRT-PCR验证分析, 4个基因在3种组织中表现相同的偏向性, 2个基因在3种组织中表现不同的偏向性, 均与RNA-Seq分析结果一致(图6)。
3 讨论
本研究表明, Illumina 双端测序技术是同时分析杂交种中基因表达水平和等位基因表达方式的一个强有力工具, 为深入研究杂种优势的分子机制提供了有价值的信息。在高丹草杂种及其亲本之间的SNP类型中, CT和GA为数量最多的类型。研究表明, 胞嘧啶甲基化可能是造成这种现象的原因[32], 发生甲基化的胞嘧啶比没有发生甲基化的胞嘧啶突变频率高, 而且甲基化的胞嘧啶发生脱氨基作用产生胸腺嘧啶T的频率高于其他自发突变的频率[33-35]。
本研究在根、茎和叶组织中的9308个等位基因表达偏向性一致的SNP中, 仅有198个存在极显著的等位基因表达偏向性, 约占21%。He等[36]发现在日本晴93-11及其杂交种中有398 (22.7%)个存在显著的等位基因表达偏向性。在玉米[37]和杨树[38]的基因中, 分别有73%和57%的基因表现出等位基因表达偏向性。翟蓉蓉等[39]发现在超级稻协优9308及其亲本中有480 (17%)个存在显著的等位基因表达偏向性。本研究中, 至少在一个组织存在等位基因表达偏向性的145个转录本中, 根、茎和叶分别有62% (36)、66% (26)和65% (32)的转录本的等位基因表达偏向白壳苏丹草, 这些转录本编码多种重要功能蛋白。在3种组织中, 与高粱11A的等位基因相比, 白壳苏丹草的等位基因更能维持它们在高丹草杂种中的活性, 并且对杂种优势做出贡献。白壳苏丹草等位基因的功能多样性也可能对高丹草杂种的优良表型起作用。在上述145个转录本中, 49个转录本(约34%)的等位基因表达在不同组织表现出不同的偏向性, 说明杂交种中的等位基因表达具有组织特异性[38, 40]。
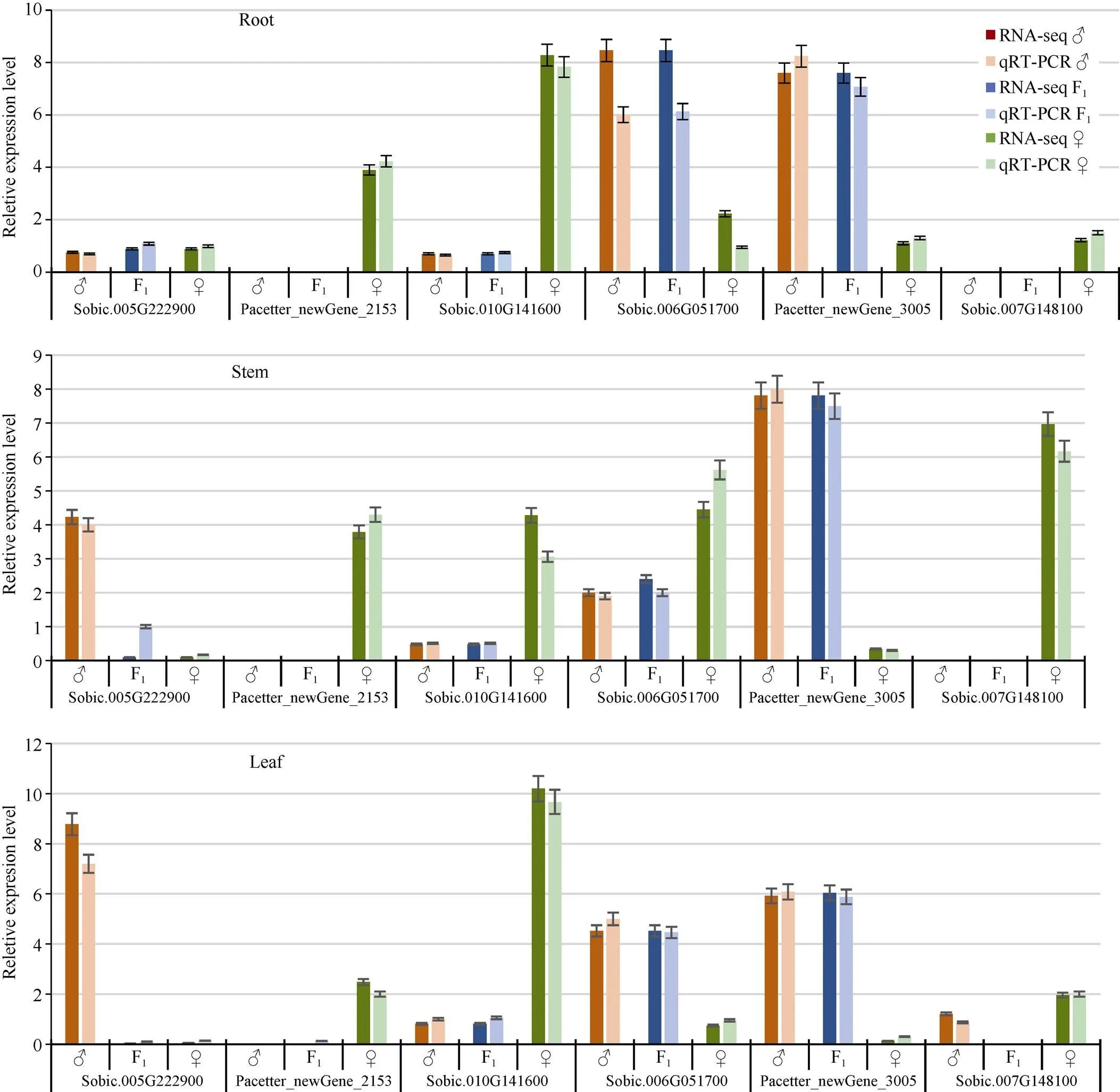
图6 高丹草杂种及其亲本3个组织中各基因表达量
顺式作用元件或反式作用因子变异均可能引起等位基因的表达变异[41]。前者可能改变启动子强度、增强子活性或转录稳定性, 而后者可能影响结构、连接和转录因子[42]。顺式和反式调控可以通过比较亲本表达水平的比值和杂交种中亲本等位基因特异性表达的比值来确定[38]。如果变异发生在顺式作用元件, 那么亲本表达水平的比值和杂交种中亲本等位基因特异性表达的比值没有差异。如果变异发生在反式作用因子, 由于杂交种中的两个亲本等位基因处于相同的亚细胞环境中, 杂交种中的双亲等位基因的表达没有差异。本研究发现, 在3种组织中,分别有79%和82%转录本的2个亲本等位基因表现出稳定的表达水平, 说明与顺式作用相比, 反式作用可能更多地影响了等位基因的特异性表达, 这些涉及抗性或者其他重要代谢反应相关转录本的等位基因所受到的差异调控可能与高丹草杂种表现出的杂种优势有关。
4 结论
共鉴定了9308个分布于整个基因组的高质量的SNP位点。Illumina测序技术是分析高丹草杂种中基因表达水平和等位基因表达方式的一个强有力工具, 为深入研究杂种优势形成的分子机制提供了有价值的信息。DNA甲基化现象可能存在于高丹草杂种及其亲本中; 与母本高粱11A相比, 父本白壳苏丹草的等位基因更能维持在高丹草杂种中的表达并对杂种优势形成产生影响; 根、茎和叶组织的高丹草杂种中亲本等位基因具有不同的表达方式, 具有组织特异性; 高丹草杂种中亲本等位基因的差异表达多数由反式作用因子调控。
[1] 詹秋文, 钱章强. 高粱与苏丹草杂种优势利用的研究. 作物学报, 2004, 30: 73–77Zhan Q W, Qian Z Q. Research of Sorghum-Sudan grass of heterosis utilization., 2004, 30: 73–77 (in Chinese with English abstract)
[2] Lu X P, Yun J F, Gao C P. Quantitative trait loci analysis of economically important traits in´hybrid., 2011, 9: 81–90
[3] Arnold M L. Natural hybridization and the evolution of domesticated, pest and disease organisms., 2004, 13: 997–1007
[4] Hegarty M J, Hiscock S J. Hybrid speciation in plants: new insights from molecular studies., 2004, 165: 411–423
[5] Rieseberg L H. Hybrid origins of plant species., 1997, 28: 359–389
[6] Rieseberg L H, Raymond O, Rosenthal D M. Major ecological transitions in wild sunflowers facilitated by hybridization., 2003, 301: 1211–1216
[7] 曹廷杰, 谢菁忠, 吴秋红, 陈永兴, 王振忠, 赵虹, 王西成, 詹克瑟, 徐如强, 王际睿, 罗明成, 刘志勇. 河南省近年审定小麦品种基于系谱和SNP标记的遗传多样性分析. 作物学报, 2015, 41: 197–206 Cao Y J, Xie J Z, Wu Q H, Chen Y X, Wang Z H, Zhao H, Wang X C, Zhan K S, Xu R Q, Wang J R, Luo M C, Liu Z Y. Genetic diversity of registered wheat varieties in Henan province based on pedigree and single-nucleotide polymorphism., 2015, 41: 197–206 (in Chinese with English abstract)
[8] Birchler J A, Auger D L, Riddle N C. In search of the molecular basis of heterosis., 2003, 15: 2236–2239
[9] 曲存民, 卢坤, 刘水燕, 卜海东, 付福友, 王瑞, 徐新福, 李加纳. 黄黑籽甘蓝型油菜类黄酮途径基因SNP位点检测分析. 作物学报, 2014, 40: 1914–1924 Qu C M, Lu K, Liu S Y, Bu H D, Fu F Y, Wang R, Xu X F, Li J N. SNP detection and analysis of genes for flavonoid pathway in yellow-and black-seededL., 2014, 40: 1914–1924 (in Chinese with English abstract)
[10] Springer N M, Stupar R M. Allelic variation and heterosis in maize: How do two halves make more than a whole?, 2007, 17: 264–275
[11] Stupar R M, Spirnger N M. Cis-transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F1hybrid., 2006, 173: 2199–2210
[12] 许家磊. 基于甘薯徐781和徐薯18转录组测序的SNP标记开发. 中国农业科学院硕士学位论文, 北京, 2015 Xu J L. Development of SNP Markers Based on Transcriptome Sequencing of Xu 781 and Xushu 18 in Sweetpotato,(L.) Lam. MS Thesis of Chinese Academy of Agricultural Sciences, Beijing, China, 2015 (in Chinese with English abstract)
[13] Pastinen T. Genome-wide allele-specific analysis: insights into regulatory variation., 2010, 11: 533–538
[14] 石璇, 王茹媛, 唐君, 李宗芸, 罗永海. 利用简化基因组技术分析甘薯种间单核苷酸多态性. 作物学报, 2016, 42: 641–647 Shi X, Wang R Y, Tang J, Li Z Y, Luo Y H. Analysis of interspecific SNPs in sweetpotato using a reduced-representation genotyping technology., 2016, 42: 641–647 (in Chinese with English abstract)
[15] 刘峰, 谢玲玲, 弭宝彬, 欧阳娴, 茆振川, 邹学校, 谢丙炎. 辣椒转录组SNP挖掘及多态性分析. 园艺学报, 2014, 41: 343–348 Liu F, Xie L L, Mi B B, Ouyang X, Mao Z C, Zou X X, Xie B Y. SNP mining in pepper transcriptome and the polymorphism analysis., 2014, 41: 343–348 (in Chinese with English abstract)
[16] Tirosh I, Reikhav S, Levy A A. A yeast hybrid provides insight into the evolution of gene expression., 2009, 324: 659–662
[17] Zhang X, Borevitz J O. Global analysis of allele-specific expression in Arabidopsis thaliana., 2009, 182: 943–954
[18] Zhai R, Feng Y, Wang H. Transcriptome analysis of rice root heterosis by RNA-Seq., 2013, 16: 19
[19] Zhang M, Li N, He W. Genome-wide screen of genes imprinted in sorghum endosperm, and the roles of allelic differential cytosine methylation., 2016, 85: 424–436
[20] 逯晓萍, 云锦凤, 肖宇红, 米福贵, 李美娜, 尹利. 高丹草(高粱×苏丹草)产量及其构成因素的QTL分析. 华北农学报, 2007, 22(4): 80–85 Lu X P, Yun J F, Xiao Y H, Mi F G, Li M N, Yin L. QTL localization and analysis on yield and related factors of Sorghum×Sudan grass., 2007, 22(4): 80–85 (in Chinese with English abstract)
[21] 逯晓萍, 刘丹丹, 王树彦, 米福贵, 韩平安, 吕二锁. 高丹草遗传效应与杂种表现预测模型. 作物学报, 2014, 40: 466–475 Lu X P, Liu D D, Wang S Y, Mi F G, Han P A, Lyu E S. Genetic effects and heterosis prediction model of×grass., 2014, 40: 466–475 (in Chinese with English abstract)
[22] 董婧, 逯晓萍, 米福贵, 王树彦, 何丽君, 韩平安, 薛春雷, 丛梦露, 李俊伟. 高丹草杂种和亲本叶片基因差异表达研究. 植物遗传资源学报, 2016, 17: 738–747 Dong J, Lu X P, Mi F G, Wang S Y, He L J, Han P A, Xue C L, Cong M L, Li J W. Relationship between differential gene expression patterns in leaves of the hybrids and their parents of., 2016, 17: 738–747 (in Chinese with English abstract)
[23] Han P A, Lu X P, Mi F G, Dong J, Xue C L, Li J K, Han B, Zhang X Y. Proteomic analysis of heterosis in the leaves of Sorghum-Sudan grass hybrids., 2016, 48: 161–173
[24] Kim D, Pertea G, Trapnell C. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions., 2013, 14: R36
[25] McKenna A, Hanna M, Banks E. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data., 2010, 20: 1297–1303
[26] Altschul S F, Madden T L, Zhang J. Gapped BLAST and PSI BLAST: a new generation of protein database search programs., 1997, 25: 3389–3402
[27] Ashburner M, Ball C A, Blake J A. Gene ontology: tool for the unification of biology., 2000, 25: 25–29
[28] Kanehisa M, Goto S, Kawashima S. The KEGG resource for deciphering the genome., 2004, 32: 277–280
[29] Tatusov R L, Galperin M Y, Natale D A. The COG database: a tool for genome scale analysis of protein functions and evolution., 2000, 28: 33–36
[30] 杨侃侃. 基于RNA-seq技术对西瓜果皮色泽差异表达基因的分析. 江西农业大学硕士学位论文, 江西南昌, 2015 Yang K K. Analysis of Genes Differentially Expressed in Watermelon Rind Color Based on RNA-seq. MS Thesis of Jiangxi Agricultural University, Nanchang, China, 2015 (in Chinese with English abstract)
[31] 杜玮南, 孙红霞, 方德福. 单核苷酸多态性的研究进展. 中国医学科学院学报, 2000, 22: 392–394 Du W N, Sun H X, Fang D F. The research development of single nucleotide polymorphism., 2000, 22: 392–394 (in Chinese with English abstract)
[32] Bransteitter R, Pham P, Scharff M D. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase., 2003, 100: 4102–4107
[33] Chodavarapu R K, Feng S, Ding B. Transcriptome and methylome interactions in rice hybrids., 2012, 109: 12040–12045
[34] Morgan H D, Dean W, Coker H A. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming., 2004, 279: 52353–52360
[35] Yebra M J, Bhagwat A S. A cytosine methyltransferase converts 5-methylcytosine in DNA to thymine., 1995, 34: 14752–14757
[36] He G M, Zhu X P, Elling A A. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids., 2010, 22: 17–33
[37] Guo M, Rupe M A, Zinselmeier C. Allelic variation of gene expression in maize hybrids., 2004, 16: 1707–1716
[38] Zhuang Y, Adams K L. Extensive allelic variation in gene expression in populus F1hybrids., 2007, 177: 1987–1996
[39] 翟荣荣. 超级稻协优9308根系杂种优势的转录组分析. 中国农业科学院博士学位论文, 北京, 2013 Zhai R R. Transcriptome Analysis of Root Heterosis in a Super Hybrid Rice Xieyou 9308 by RNA-Seq. PhD Dissertation of Chinese Academy of Agricultural Sciences, Beijing, China, 2013 (in Chinese with English abstract)
[40] Springer N M, Stupar R M. Allelic-specific expression patterns reveal biases and embryo-specific parent-of-origin effects in hybrid maize., 2007, 19: 2391–2402
[41] Wittkopp P J, Haerum B K, Clark A G. Evolutionary changes in cis and trans gene regulation., 2004, 430: 85–88
[42] Shen Y, Catchen J, Garcia T. Identification of transcriptome SNPs between Xiphophorus lines and species for assessing allele specific gene expression within F1interspecies hybrids., 2012, 155: 102–108
Analysis of SNP and Allele-specific Expression in Transcriptome of×and Their Parents
DONG Jing1, LU Xiao-Ping1,*, ZHANG Kun-Ming1, XUE Chun-Lei1, and ZHANG Rui-Xia2
1Agronomy College, Inner Mongolia Agricultural University, Huhhot 010019, Inner Mongolia, China;2Huhhot Seed Management Station, Huhhot 010020, Inner Mongolia, China
Taking root, stem and leaf tissues ofhybrids and their parents as test materials, Illumina Hiseq 2000 was used to analyze the transcriptome to explore the relationship between single nucleotide variation and heterosis in the hybrids ofand their parents. About 58 000 SNP loci were detected from the sequencing samples with an average length of 58 122 160 bp. The number of genic SNP was significantly more than that of intergenic SNP. The frequency of SNP was 1/741 bp, and the conversion ratio of the average conversion was 1.00:1.53. Among all the types of variation, C/T and G/A had the highest frequency. After screening, 198 (21%) extremely significant biased alleles were expressed in bias SNP, and 65% of them were biased towards paternal white shell, and many of the transcriptional copies with high level gene expression in the white shellwere also expressed in thehybrid. The two parental alleles with 79%, 78%, and 82% transcripts showed a stable level of expression in the three tissues. It is suggested that the trans-acting may affect the specific expression of the allele more than the-acting. Six highly-biased SNP-unigene alleles were selected for qRT-PCR validation. The differential gene expression pattern of these genes was consistent with that of RNA-Seq analysis. Illumina sequencing technology was used to study allelic expression in this study, provides a basis for heterosis analysis ofand also a theoretical reference for related studies of other forage crops.
; heterosis; transcriptomics; single nucleotide polymorphism; functional annotion
2017-12-21;
2018-07-20;
2018-07-25.
10.3724/SP.J.1006.2018.01809
通信作者(Corresponding author):逯晓萍, Email: lxp1960@163.com
Email: 984012971@qq.com
本研究由国家自然科学基金项目(31160302, 31460375)和呼和浩特市科技计划项目(2012-重-计-8-2)资助。
This study was supported by the National Natural Science Foundation of China (31160302, 31460375) and the Science and Technology Plan Projects of Hohhot (2012-major-plans-8-2).
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20180724.1631.006.html
