高斯计算中分子结构独特性的判断
2018-11-28王海燕崔文超丁益宏
王海燕, 崔文超, 于 爽, 丁益宏
(1. 吉林师范大学 计算机学院, 吉林 四平 136000; 2. 吉林大学 理论化学研究所, 长春 130021)
化学反应过程中涉及大量的中间体和过渡态, 在确定这些中间体和过渡态过程中需构建数量更多的中间体和过渡态作为化学结构计算程序(如高斯软件包)的输入结构. 通过高斯计算获得有效结构, 进一步确定低能结构或有效的化学反应通道. 由于存在重复结构, 因此需要将其去除, 留下独特的结构. 分子结构产生有多种途径, 如通过假设或借助计算机算法产生初始结构, 并判断产生的结构是否合理, 以及是否存在结构的独特性等. 在判断结构独特性的过程中, 可通过以下过程实现: 将初始结构输入高斯计算程序中进行深层次计算、 优化, 最终得到一个终止结构. 但相同的终止结构会存在于大量的高斯计算输出文件中. 在去重过程中, 简单结构可通过肉眼分辨, 但若分子中原子数较多则会导致分子的空间构型复杂, 去重操作困难.
计算化学的主要目标是利用有效的数学近似以及电脑程序计算分子的性质(如总能量、 偶极矩、 四极矩、 振动频率、 反应活性等), 并解释一些具体的化学问题[1-2]. 在计算化学应用领域, 常将已有的电脑程序和方法针对特定的化学问题进行研究, 如在构建分子结构过程中, 可利用计算机辅助方式; 在结构搜索中, 需判断结构的独特性, 如C2Si2Xq(五原子高周期14族平面四配位化合物)[3-4]、 C2Al4H4[5]、 铝氢团簇和氮杂环卡宾等[6], 存在大量结构相同的分子, 针对计算机程序产生的大量结构文件, 若不去重, 则会导致大量冗余工作. 而仅凭能量相等判断分子结构的独特性易出现失误. 针对上述问题, 本文将计算机辅助方式应用于结构搜索子方向, 提出一种自动化结构独特性判断STRUauto算法, 该算法借助能量和键长的迭代比较, 依次完成分子结构的批量去重操作, 可避免常规分子结构独特性判断的误差, 准确得到分子的异构体. 为验证STRUauto算法的优势, 先对C2Si2Xq和C2Al4H4两类典型的分子结构进行实验测试, 再对C2Si2Xq中的六类典型体系进一步实验. 分别考虑常规状态下和应用STRUauto状态下的多项典型技术指标比对, 实验结果表明, STRUauto算法能有效提升理论化学中结构搜索问题的效率, 其准确率和可信度良好.
1 自动化结构独特性判断算法
1.1 问题描述 两个具有相同原子数目的分子其结构可能存在差异, 如C2Al4H4可排列成多种分子结构. 研究表明, 当原子数目增加时, 分子结构数目会成倍增加. 除通过肉眼和经验值判断C2Al4H4分子可能存在独特的结构, 目前还有许多辅助性的识别方法, 如借助总能量值判断. 计算出分子结构后, 可计算出一个能量. 总能量值相近的分子, 易误认为结构相同. 一些能量相近但结构不同的实例列于表1, 表1中实例的分子结构构型如图1所示.
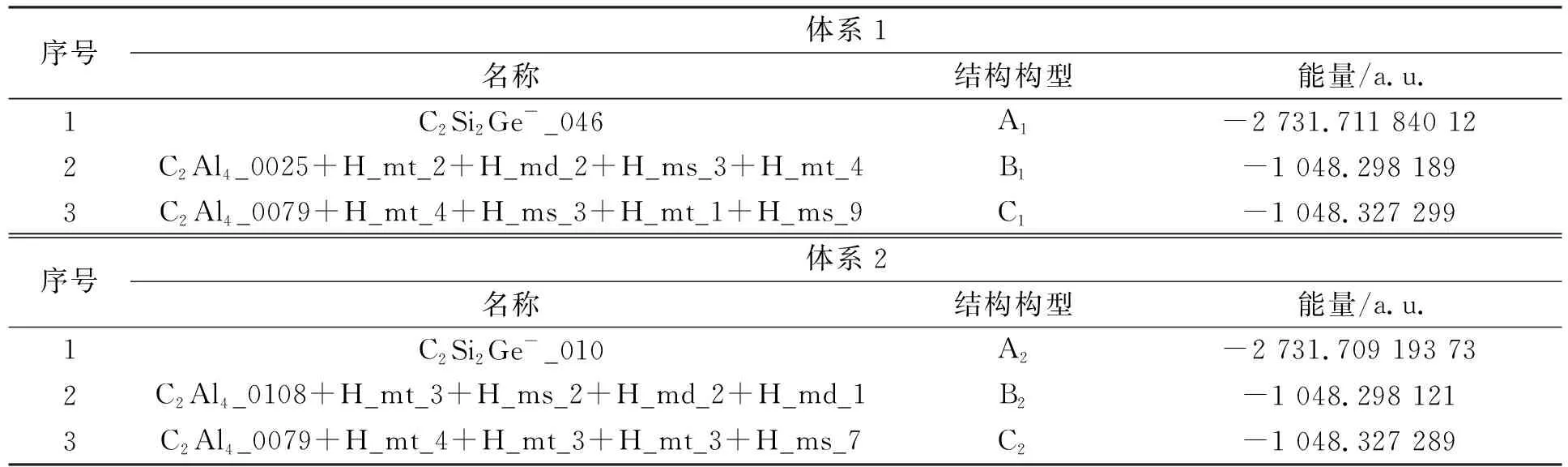
表1 能量相近但结构不同实例Table 1 Examples of similar energy but different structures

图1 表1中实例的分子结构构型Fig.1 Molecular structural configuration of examples in tabel 1
由表1可见: 在C2Si2Ge-体系中, 第一组中C2Si2Ge-_046和C2Si2Ge-_010的能量接近, 其能量差仅为0.002 646 39 a.u., 但其结构构型完全不同; C2Al4_0025+H_mt_2+H_md_2+H_ms_3(简写)和C2Al4_0108+H_mt_3+H_ms_2+H_md_2(简写)的能量更接近, 能量差仅为0.000 068 a.u., 但其结构构型完全不同; C2Al4_0079+H_mt_4+H_ms_3+H_mt_1(简写)和C2Al4_0079+H_mt_4+H_mt_3+H_mt_3(简写)的能量差仅为0.000 01 a.u., 结构构型相似, 但甲基的转动位置不同, 因此二者属于两种不同结构. 所以仅凭能量相等或能量差小于某个阈值判断分子结构的独特性会得到错误结论.
1.2 算 法 通常可由分子中原子个数及各原子的空间位置判断一种分子的结构独特性. 但对于原子个数相同且空间位置相似的分子, 即使加入经验值, 判断也很困难. 因此, 需要一种自动化的结构独特性判断算法, 以提高判别的准确度和效率. 研究表明, 分子总能量是判别结构独特性的一个关键因素. 若最初按分子结构的总能量进行判断, 则可先区分出一些异构体(中间体), 即能量不同, 其结构不同, 进而确定部分分子的独特性. 其次, 分子中各原子间的键长是另一个关键因素. 总能量相同的分子, 结构内部原子间键长不同也会导致两种分子的异构. 基于此, 本文提出一种自动化判断两种分子结构独特性的STRUauto算法, 算法描述为: 1) 预处理过程; 2) 判断总能量的一致性; 3) 判断键长的一致性; 4) 去重或确定分子结构独特性. 其工作流程如图2所示.
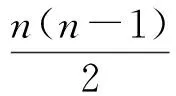
在判断分子总能量一致性的过程中, 先通过化学结构计算程序(如高斯软件包等)确定所有中间体总能量, 再计算中间体之间的能量差, 这是决定异构体独特性的首要因素. 考察能量差需预先设定一个阈值, 实验结果表明, 阈值应设为9×10-5a.u., 若2种分子结构的能量差大于阈值, 则2个结构必不同, 进而确定为独特结构; 若能量差小于9×10-5a.u., 则需进一步判断. 在进一步判断过程中, 主要决定因素是原子与原子间的键长以及2个原子的类型. 键长决定2个结构的内部距离, 内部距离构成分子的空间结构. 由于相同的键长值可能对应不同的原子组合, 但不需要判断键长的一致性, 因此, 需要判断是否对键长升序排序. 对于相同的原子组合, 才将这些距离按升序规则排序.
2 常规方法与自动化方法比较
为证明STRUauto自动化结构独特性判断算法的优越性, 将STRUauto算法与常规异构体判断方法进行对比. 实验环境为: Intel(R) Xeon(R), X5570, 2.93 GHz处理器, 32 GB内存[7].
为验证STRUauto算法的效率[8], 选取典型的分子组合C2Si2Xq和C2Al4H4为研究对象, 分别研究具有相同原子组合的分子结构独特性, 其技术指标包括初始结构数目、 异构体数目、 异构体时间、 去重比例和准确率. 通过上述指标比较常规方法与自动化判断STRUauto算法的差异, 并对上述指标进行统计分析.
2.1 结构去重结果比较 C2Si2Xq和C2Al4H4的初始结构分别为24 892,4 372个, 用STRUauto算法找出异构体的数目分别为3 442,552个. C2Si2Xq体系内的结构去重结果列于表2. 由表2可见, 由于STRUauto算法考虑了结构总能量、 原子与原子间的键长以及2个原子类型等因素, 因此去除了初始结构中的重复结构, 有效得到了相应数目的异构体.
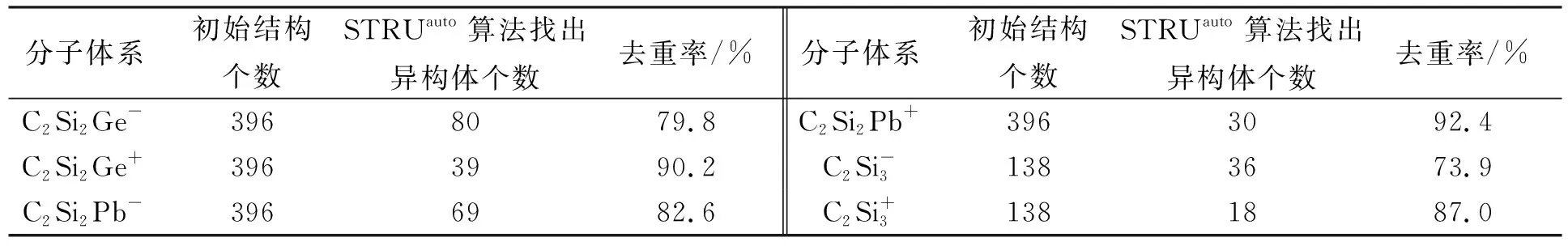
表2 C2Si2Xq体系内的结构去重结果Table 2 Deduplication results of structure in C2Si2Xq system
以C2Si2Ge-为例对数据进行统计分析. C2Si2Ge-体系的初始结构有396个, 经程序去重, STRUauto找到same文件夹(所有存在相同结构的文件集合)中的结构有316个, 最终确定80个异构体, 去重率为79.8%. 经确认, 找出的80个结构均为独特结构, 即发现异构体的准确率为100%. 表2中所有分子体系找出的结构均为独特结构, 发现异构体的准确率均为100%.
为验证STRUauto算法在异构体去重方面的优势, 将自动化结构独特性判断STRUauto算法与常规人工分子结构独特性判断方法在去重率方面进行比较. 实验结果表明, 常规方法的去重率均小于50%, STRUauto算法的去重率均大于70%.
综上所述, 本文利用计算机辅助分子异构体构建了STRUauto算法, 结果表明: 在结构搜索过程中, 即使2个结构的能量相同, 其分子结构也可能不同, 因此需要进一步借助其他辅助因素判断, 最合适的辅助因素是原子间的键长以及2个原子的类型; 当结构能量差值较小时, 通过2组n(n-1)/2个键长的比较可确定2个结构是否相同. STRUauto算法针对结构搜索体系中原子个数增多与筛选准确率之间的矛盾, 较精准地完成了化学结构搜索的去重问题. 当分子总能量、 原子数和键长均相同时, 可排除存在异构体的可能. 通过STRUauto算法与常规方法的数据比较, 证明其准确率和效率均有较大提高.
