二代测序在家族遗传性高危胃肠肿瘤筛查中的应用*
2018-11-28
消化道肿瘤是世界范围内高发的恶性肿瘤。中国人群消化道肿瘤的发病率逐年升高,其中胃癌发病率位居第3位,结直肠癌位居第5位[1]。绝大多数胃癌为散发性病例,但约10%患者具有家族聚集特征,其中约1/3被认为具有遗传背景。遗传性弥漫型胃癌主要与CDH1的胚系突变有关,病理类型多为弥漫型和(或)印戒细胞癌;除了胃癌,小叶性乳腺癌和结直肠癌等肿瘤的发生率也明显升高[2]。对于遗传家系中CDH1基因突变携带者终生胃癌发病风险高达60%~80%[3]。同样在结直肠癌中,约30%患者具有家族遗传特征,其中有明确致病因素的遗传性结直肠癌约占5%~6%[4]。目前,发病率最高、研究相对系统的Lynch综合征约占全部结直肠癌的3%,由错配修复基因的胚系突变导致,其携带者具有早发结直肠癌和易患第二原发肿瘤的特点[5-7]。对于携带肿瘤遗传相关基因人群进行及时的诊断和规范的随访监测,有助于降低该特定人群肿瘤发生率及死亡率,具有明确的一、二级预防价值。
对遗传性非息肉病性结直肠癌(hereditary non⁃polyposis colorectal cancer,HNPCC)来说,虽然修正的“百慕大标准”明显提高了诊断的敏感度,但仍有20%~30%患者会被遗漏[7]。遗传性弥漫性胃癌更因国内认知及研究匮乏,容易被临床忽视。目前,关于遗传性消化肿瘤筛查的结果显示,50%甚至更多符合临床诊断的患者或家系无法找到明确的分子致病机制。其原因包括:1)目标基因新的致病性突变未被发现;2)由于临床表现多样和肿瘤家族史不典型,其临床诊断的遗传综合征与遗传致病基因不符。因此,传统的表型特异性单基因检测模式已难以满足当前遗传性消化道肿瘤的诊疗需求。随着测序技术的进步[8],对遗传性肿瘤多基因平台的同时性检测,极大地推动了低外显率及未知突变的发现,因此运用二代测序的方法对遗传性消化道肿瘤进行筛查具有广阔的临床应用前景。
本文拟通过发病年龄、家族史、病理类型、肿瘤微卫星状态等遗传性消化道肿瘤的危险因素,筛选高危人群,通过二代测序的方法同时性检测42个遗传性肿瘤相关基因,探索对遗传性消化道肿瘤高危患者进行遗传筛查的意义及不同危险因素在患者筛选中的价值。
1 材料与方法
1.1 病例资料
选取2016年3月至2016年4月收治于北京大学肿瘤医院的322例结直肠癌及胃癌患者,筛选满足遗传性胃肠肿瘤的高危人群,入组患者需满足下述1项:1)患者年龄≤30岁,不论有无家族史;2)患者年龄≤35岁,不论有无家族史;特殊病理类型:印戒细胞癌或黏液腺癌;3)患者年龄≤50岁;且≥1位一级亲属中发生恶性肿瘤;4)≥2位一、二级亲属发生恶性肿瘤,其中至少1位是一级亲属;5)患者本身≥2种恶性肿瘤,且患第1种恶性肿瘤时年龄≤50岁;6)组织标本微卫星不稳定(microsatellite instability,MSI)或错配修复蛋白缺失。共筛选79例满足入组条件患者,排除临床资料不完善及样本不完整者,共入组患者25例。本研究经过北京大学肿瘤医院伦理委员会审批(编号:2016YJZ52)。
1.2 方法
1.2.1 标本的获取 入组患者抽取外周血5 mL,使用 Maxwell®RSC DNA FFPE Kit试剂和 Maxwell®RSC自动提取仪(均购自美国Promega公司)按照说明书标准操作流程进行全血样本DNA提取。提取的DNA使用NanoDropTM2000(购自美国Thermo Fisher公司)进行检测,使用Qubit®3.0荧光定量仪(购自美国Thermo Fisher公司)进行定量。
1.2.2 胚系基因检测 所有患者均进行了42个遗传性肿瘤相关基因的胚系检测,包括:APC、ATM、BLM、BMPR1A、BRCA1、BRCA2、CDH1、CDKN2A、CFTR、CHEK2、CTNNA1、DDB2、EPCAM、ERCC1、ERCC2、ERCC3、ERCC4、ERCC5、FANCC、FANCG、GALNT12、GREM1、MLH1、MLH3、MSH2、MSH6、MSR1、MUTYH、PALB2、PMS2、POLD1、POLE、POLH、PRSS1、PTEN、SMAD4、SPINK1、STK11、TGFBR2、TP53、XPA、XPC。
1.2.3 技术及数据分析 全外显子组高通量测序技术基于美国Agilent公司的SureSelect全外显子组捕获技术对人类全部已知基因的外显子区域进行捕获。采用SureSelect Human All Exon V5+UTRs Kit试剂盒。捕获后的DNA序列采用美国Illumina公司的HiSeq X10高通量测序平台的标准文库构建和测序流程进行测序测序,对高通量数据运用GATK(Genome Analysis Toolkit,https://software.broadinstitute.org/gatk/)标准方法流程进行分析,基因突变位点的临床意义主要参照ClinVar数据库[9]。
2 结果
2.1 一般资料
本研究共入组25例患者,其中男性占48%(12/25),≤50岁的患者占72%(18/25),结直肠癌和胃癌患者分别占76%(19/25)和24%(6/25),肿瘤组织有黏液腺癌和印戒细胞癌成分的占42%(10/25),绝大多数患者具有肿瘤家族史80%(20/25),2例患者自身患有2种恶性肿瘤,免疫组织化学显示错配修复基因表达缺失(mismatch repair-deficient,dMMR)的患者占32%(8/25),错配修复蛋白表达正常(mismatch repair-proficient,pMMR)的患者占28%(7/25),微卫星状态未知的患者占40%(10/25)。
2.2 突变患者临床分子特征
入组的25例患者中,6例患者检测到病理性胚系突变,总发生率为24%。其中50%(3/6)患者肿瘤组织表现为错配修复蛋白缺失,83%(5/6)患者发病年龄≤50岁且具有恶性肿瘤家族史(表1)。
1例MYH相关性息肉病患者[10(MUTYH,c.467G>A:p.Trp156Ter)临床诊断为胃肠息肉综合征及早发结肠癌,该患者2例一级亲属均在35岁诊断为结直肠癌,1例一级亲属因结肠息肉行全结肠切除术。1例APC(c.3126_3130del:p.I1042fs)移码突变患者临床表现出家族性腺瘤性息肉病的特征,并且其2例一级亲属及1例二级亲属均于50岁前诊断出结直肠癌。4例患者携带遗传性结直肠癌相关基因突变,其中1例MLH1(c.2041G>A:p.Ala681Thr)错义突变为Lynch综合征明确的致病突变[11];1例MLH3(c.2221G>T:p.Val741Phe)胚系突变的患者40岁诊断为直肠腺癌,该患者的哥哥存在MLH3相同位点的突变,并且临床表现为小肠腺鳞癌及肝癌,进一步提供家系验证;1例TGFBR2突变(c.C944T:p.T315M)患者41岁诊断为升结肠中分化腺癌伴黏液腺癌,并且其降结肠及小肠均发现腺癌病灶;1例患者检测出MSH6(c.C1387T:p.Q463X)的胚系无义突变,且该患者肿瘤组织MSH6蛋白表达缺失。
全部入组患者满足的筛选条件及致病突变患者在其中所占的比例,见表2。全部患者中满足年龄≤50岁且≥1例一级亲属发生恶性肿瘤的比例最高(52%),满足≥2例一级/二级亲属发生恶性肿瘤,其中至少1例是一级亲属的患者11例(44%),错配修复蛋白缺失的患者占32%(8/25)。
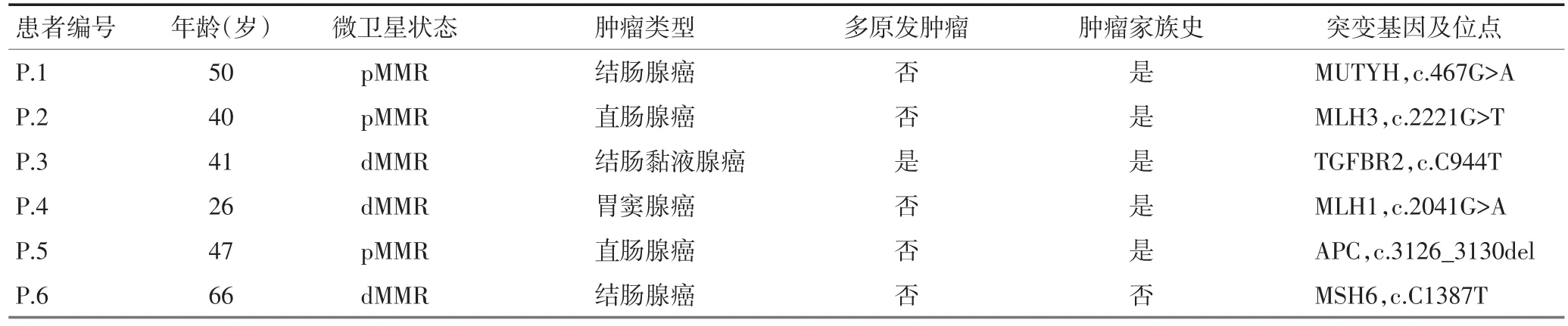
表1 具有致病性突变患者的临床特征及突变基因

表2 满足不同入组标准的全部患者人数及致病突变患者的比例
3 讨论
遗传性胃肠道肿瘤综合征是一组非表型特异性的疾病,以家系中个体肿瘤遗传易感为特征[12]。近年来,随着对肿瘤遗传综合征认知的进步和基因检测技术的进步,遗传性肿瘤的临床诊断和致病机理研究也得到了发展。如遗传性结直肠癌分类的细化及新的致病基因和突变位点的发现[6,13-15],以及遗传性慢性胃癌与CDH1基因突变的关系及临床诊断标准的改进等。中国对于遗传性胃肠道肿瘤的研究虽然起步较晚,但是相关研究也提示国内外遗传性肿瘤发生谱不同的可能[16]。最近,Pearlman 等[17]针对450例早发结直肠患者应用二代测序进行25个遗传相关基因的筛查,结果显示72例患者具有遗传相关基因的胚系致病突变。接近16%的高突变率提示二代测序在遗传性肿瘤综合征研究中的应用价值。
由于遗传性肿瘤综合征的临床表现多样和肿瘤家族史不典型,其临床诊断与遗传致病基因不符的情况较为常见,传统的表型特异性单基因检测模式已不适用于当前遗传性消化道肿瘤的诊疗需求。本研究结合目前研究相对成熟的遗传性弥漫性胃癌及Lynch综合征[11]的临床诊断标准,以发病年龄、特殊病理类型、恶性肿瘤家族史等因素作为高危人群的筛选条件,运用二代测序技术检测42个肿瘤遗传相关基因的全外显子,发现24%患者携带致病性胚系突变。其中3例分别为具有明确致病性MLH1、APC和MUTYH胚系突变,并且这些突变与患者的临床病理学特征相符。另外,1例携带MLH3 c.2221G>T突变患者40岁诊断为直肠腺癌且具有Lynch综合征相关恶性肿瘤家族史,虽然该基因突变位点在ClinVar数据库中显示致病性有争议,但患者的一级亲属(哥哥)临床诊断小肠腺鳞癌及肝脏神经内分泌肿瘤,同时检测出MLH3相同位点的突变,提供了家系验证。并且既往研究也报道了2例携带MLH3 c.2221G>T突变的Lynch相关肿瘤家系[18-19],第一个家系中先证者为1例71岁诊断结直肠癌的男性患者,2例一级亲属分别于57岁和61岁诊断为乳腺癌及胃癌,3例患者均存在MLH3 c.2221G>T突变。第二个家系先证者为1例子宫内膜癌女性患者,本身及2例一级亲属均存在MLH3 c.2221G>T突变,且其中1例一级亲属也诊断为子宫内膜癌。1例患者检测出TGFBR2胚系突变(c.C944T:p.T315M),既往报道该突变位点存在于微卫星稳定的HNPCC家系中[20]。1例肿瘤组织MSH6蛋白表达缺失的患者,检测出MSH6的无义突变。本研究结果显示,即使通过比较宽松的条件筛选遗传性消化道肿瘤的高危患者,其中病理性突变的发生率仍可达25%,较以往整体人群5%的比例明显提高。胚系突变检出率的提高将有助于对携带者进行早期的诊断和调整相应的随访监测,从而最终降低该特定人群肿瘤发生率及死亡率,具有重要的一、二级预防价值。
遗传性综合征常具有多种肿瘤易感倾向,如本研究中携带MLH1致病突变的患者诊断为胃窦腺癌,临床特征并不符合“阿姆斯特丹标准”;既往报道乳腺癌的家系符合遗传性弥漫型胃癌的基因突变特点,使得针对单一肿瘤部位的遗传筛查可能造成偏差及漏诊[21]。因此,对遗传性肿瘤高危人群筛选时可考虑不再对肿瘤原发部位进行严格限定。关于不同的筛选条件在遗传性消化道肿瘤中的价值,研究初步显示多原发肿瘤病史、微卫星不稳定状态及恶性肿瘤家族史在筛查遗传性消化道肿瘤中的权重较大。携带致病突变的患者中,50%患者具有恶性肿瘤家族史,50%患者肿瘤组织错配修复蛋白表达缺失。本研究显示,患者的家族史及肿瘤组织微卫星状态在遗传性肿瘤综合征临床诊断中具有重要作用。虽然进行更严格的年龄限定似乎并未增加遗传性肿瘤的检出率,但是携带致病性胚系突变的患者发病年龄均≤50岁,仍符合遗传性肿瘤综合征早发的特点。同样,进行更严格的年龄限定似乎不如综合患者的年龄、家族史和微卫星状态更具有临床价值。
传统的表型特异性单基因检测模式不再适用于遗传性肿瘤综合征的诊断,近些年二代测序技术在遗传病中应用的重要性也逐渐突出,尤其是全外显子测序或针对多个遗传相关基因谱的测序为遗传性消化道肿瘤的诊断和研究提供了大量信息。对于具有遗传性肿瘤家系进行二代测序,能够发现传统检测方法未发现和(或)既往尚未报道的新致病性基因突变[13,22]。同时,在具有消化道肿瘤遗传性高危家系中,利用二代测序对多个相关遗传基因检测,可发现部分高危家系存在目前临床标准无法诊断致病性的遗传基因突变[23-24]。多基因平台的同时性检测,可以降低漏诊率,一定程度上解决表型与基因型不一致的情况。如MLH3、TGFBR2与遗传性非息肉病性结直肠癌的关系在本研究中得到了进一步验证。
本研究具有样本量小的局限性,并且检测平台均对目前已知的基因进行检测,并不能发现新的致病基因。二代测序面临的另外一个问题是检测出大量意义未明的基因突变,目前尚缺乏统一的方法明确其意义。如何正确地解读这些数据,需要多专业的交叉合作。综上所述,通过高危因素的筛查,富集遗传性消化道肿瘤的高危人群,进行多基因平台的同时性检测具有明确的临床意义及良好的应用前景。
致谢:感谢领星生物科技有限公司提供技术支持,对本研究中的患者进行全外显子组高通量测序及相关基因分析。
