GC-MS/MS法测定浙贝母中联苯菊酯等3种农药残留量
2018-11-24方艺霖余敏灵
方艺霖,余敏灵
(四川省乐山市食品方法检验检测中心,乐山 614000)
近年来,随着中药材产业化发展,农药污染问题日益严重。中药材种植及中药饮片加工、储存过程中农药的不当使用,均会影响中药饮片的质量,严重危害人体健康。《中国药典》2015年版四部收载了多种农药残留量的气相色谱检查方法[1]。目前对农药残留量研究报道的文献多以气相色谱法、液相色谱法、气相色谱串联质谱法及液相色谱串联质谱法为主[2-11],但都存在对照品获得困难、操作繁琐或多指标检测成本高等问题。浙贝母收载于《中国药典》2015年版一部,具有清热、化痰、止咳、解毒、散结和消痈的功效[12],目前关于浙贝母的文献报道多以药理研究为主,还未见应用GC-MS/MS法测定其多种农药残留量的文献报道。本文应用GC-MS/MS法,根据色谱保留时间和质荷比信息对样品进行定性分析,以联苯菊酯二级离子扫描色谱峰(181.1/166.1)面积作为参照,计算其与甲氰菊酯二级离子扫描色谱峰(265.1/210.1)面积和溴氰菊酯二级离子扫描色谱峰(252.9/93.0)面积的校正因子,建立GS-MS/MS法测定浙贝母中联苯菊酯等3种农药残留量的一测多评法,以期为GC-MS/MS法测定中药中农药残留量提供一测多评[13-14]研究思路。
1 仪器与试药
1.1仪器 GCMS-TQ8030气质联用仪,Lab Solutions/GCMS solutions 4.20版本工作站(岛津中国科技有限公司);XS205 DualRange 型电子天平(瑞士梅特勒-托利多仪器有限公司)
1.2试药 浙贝母样品购于乐山中药材有限公司,按照《中国药典》2015年版鉴定为百合科植物浙贝母FritillariaThunbergiiMiq.的干燥鳞茎,样品批号分别为20170901,20170902和20170903。联苯菊酯、甲氰菊酯和溴氰菊酯对照品溶液(规格:100 μg·mL-1,批号分别为:GSB05-2333-2016,GSB05-2306-2016和GSB05-2310-2016,农业部环境监测保护研究所);Cleanert TPH净化柱(天津博纳艾洁尔公司);乙腈、丙酮为色谱纯,氯化钠、无水硫酸钠为优级纯,水为纯化水。
2 方法与结果
2.1测定原理 以联苯菊酯为对照品测定样品中3种农药残留的原理是利用样品色谱图中联苯菊酯、甲氰菊酯和溴氰菊酯的色谱峰相对保留时间进行定位以及二级离子碎片图谱碎片离子峰和丰度比对联苯菊酯、甲氰菊酯和溴氰菊酯色谱峰进行定性分析。在定量分析时,通过精密量取联苯菊酯、甲氰菊酯和溴氰菊酯对照品溶液各适量,配制成不同质量浓度的混合溶液,按照设定条件进样,记录色谱图,绘制进样量对其峰面积的回归曲线,以联苯菊酯回归曲线斜率与甲氰菊酯和溴氰菊酯回归曲线斜率的比计算校正因子(fs/k=Ks/Kk),通过联苯菊酯回归曲线计算联苯菊酯含量,通过校正因子及联苯菊酯回归曲线计算甲氰菊酯和溴氰菊酯的含量。
2.2溶液的制备
2.2.1对照品储备液 精密量取联苯菊酯、甲氰菊酯和溴氰菊酯对照品溶液各1 mL,置于10 mL量瓶中,加丙酮稀释至刻度,摇匀,作为对照品储备液(联苯菊酯、甲氰菊酯和溴氰菊酯质量浓度均为10 μg·mL-1)。
2.2.2样品溶液和空白溶液 取浙贝母(批号20170901)50 g,粉碎,过三号筛,精密称取粉末1.017 9 g,置于50 mL离心管中,加入15 mL乙腈,以15 000 r·min-1匀浆提取1 min,加入氯化钠2 g,再匀浆提取1 min,以4 200 r·min-1离心5 min,取上清液,置于150 mL鸡心瓶中,在离心管中加入15 mL乙腈,按照上述方法重复匀浆提取1次,合并提取液,置于40 ℃水浴中旋转蒸发至1 mL。在Cleanert TPH固相萃取柱上加入2 cm高的无水硫酸钠,用10 mL正己烷-丙酮(6∶4)预洗后,将样品浓缩液移入柱中,用25 mL正己烷-丙酮(6∶4)洗脱,收集洗脱液,于40 ℃水浴旋转浓缩至近干,用丙酮定容至1 mL,用0.2 μm微孔滤膜过滤,即得;除称取样品粉末外,按照上述操作制备空白溶液。
2.2.3样品加标溶液 精密称取2.2.2项下粉碎的浙贝母粉末1.027 7 g,按照2.2.2项下方法制备样品溶液。从置于50 mL离心管中起到收集洗脱液于40 ℃水浴旋转浓缩至近干,用丙酮移至1 mL量瓶中,另取2.2.1项下制备的对照品储备液1 mL,置于10 mL量瓶中,用丙酮稀释至刻度,摇匀,精密量取此溶液5 μL,置于上述样品溶液中,用丙酮定容至刻度,摇匀,用0.2 μm微孔滤膜过滤,即得。
2.3测定条件
2.3.1气相色谱条件 采用Rxi-5SilMS毛细管柱(30 m×0.25 mm,0.25 μm);进样口温度:250 ℃;线速度:47.2 cm·s-1;程序升温:50 ℃保持1 min,以25 ℃·min-1速率升至125 ℃,再以10 ℃·min-1速率升至300 ℃,保持15 min;进样体积:1 μL;载气:高纯氦气(质量分数>99.999%)。
2.3.2质谱条件 离子源温度:200 ℃;接口温度:250 ℃;溶剂延迟时间:5 min;碰撞气氩气(质量分数>99.999%);电离方法见表1。
表1电离方法
Tab.1 The ionization mode
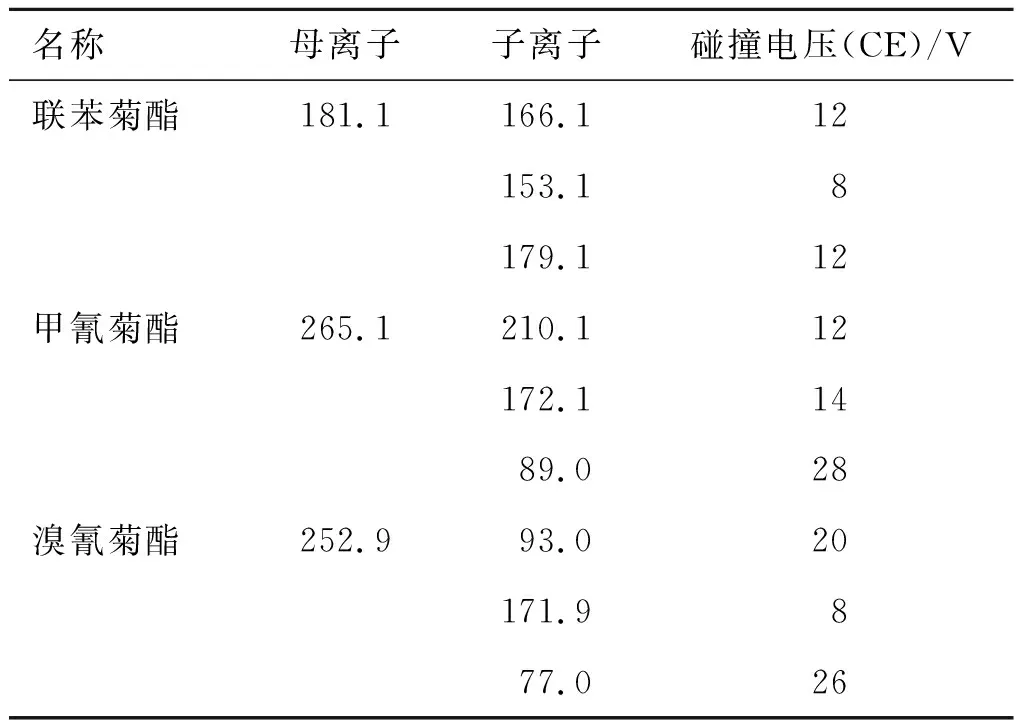
名称母离子子离子碰撞电压(CE)/V联苯菊酯181.1166.112153.18179.112甲氰菊酯265.1210.112172.11489.028溴氰菊酯252.993.020171.9877.026
2.4图谱解析 精密量取2.2.1项下制备的对照品储备液25 μL,置于5 mL量瓶中,加丙酮稀释至刻度,摇匀,作为对照品溶液。取2.2项下制备的对照品溶液、样品溶液、样品加标溶液和空白溶液分别进样,按照2.3项下测定条件进行测定,记录色谱图,在对照品溶液二级离子扫描色谱图中有联苯菊酯二级离子扫描(181.1/166.1)、甲氰菊酯二级离子扫描(265.1/210.1)和溴氰菊酯二级离子扫描(252.9/93.0)色谱峰,联苯菊酯、甲氰菊酯和溴氰菊酯保留时间分别为17.514,17.695和21.783 min,在空白溶液色谱图和样品溶液色谱图中无上述色谱峰,在样品加标溶液色谱图中有与对照品溶液色谱图中保留时间一致的联苯菊酯、甲氰菊酯和溴氰菊酯二级离子扫描色谱峰,见图1;对照品溶液和样品加标溶液的二级离子碎片图谱中联苯菊酯在153.1,166.1和179.1处(丰度比为5∶100∶19,以166.1为参比)、甲氰菊酯在为89.0,172.1和210.1处(丰度比为40∶14∶100,以210.1为参比)、溴氰菊酯在77.0,93.0和171.9处(丰度比为22∶100∶84,以93.0为参比)均有二级离子碎片峰,见图2。

图1二级离子扫描色谱图
A.对照品溶液;B.样品溶液;C.样品加标溶液;D.空白溶液;1.联苯菊酯;2.甲氰菊酯;3.溴氰菊酯。
Fig.1.The secondary ions scanning chromatograms
A.standard solution;B.sample solution;C.sample solution of adding standard;D.blank solution;1.bifenthrin;2.fenpropathrin;3.deltamethrin.
2.5线性关系考察 精密量取2.2.1项下制备的对照品储备液5,10,20,25,30,40和50 μL,分别置于7个5 mL量瓶中,加丙酮稀释至刻度,摇匀。按照2.3项下方法测定,以联苯菊酯、甲氰菊酯和溴氰菊酯的二级离子扫描峰(181.1/166.1,265.1/210.1和252.9/93.0)面积为纵坐标(y)、进样量(ng)为横坐标(x),进行线性回归,计算回归方程,结果联苯菊酯、甲氰菊酯和溴氰菊酯3种成分的回归方程分别为y1=10 388 461x1-236,y2=523 142x2-283和y3=520 881x3-144;三者的线性范围均为0.01~0.1 ng;相关系数r值均为0.999 7。
2.6f值和t值的计算与耐受性考察
2.6.1f值和t值的测定 甲氰菊酯与联苯菊酯色谱峰峰面积的校正因子f甲/联=523 142/10 388 461=0.050 4,相对保留时间t甲/联=17.695/17.514=1.010 3;溴氰菊酯与联苯菊酯色谱峰峰面积的校正因子f溴/联=520 881/10 388 461=0.050 1,相对保留时间t溴/联=21.783/17.514=1.243 7。
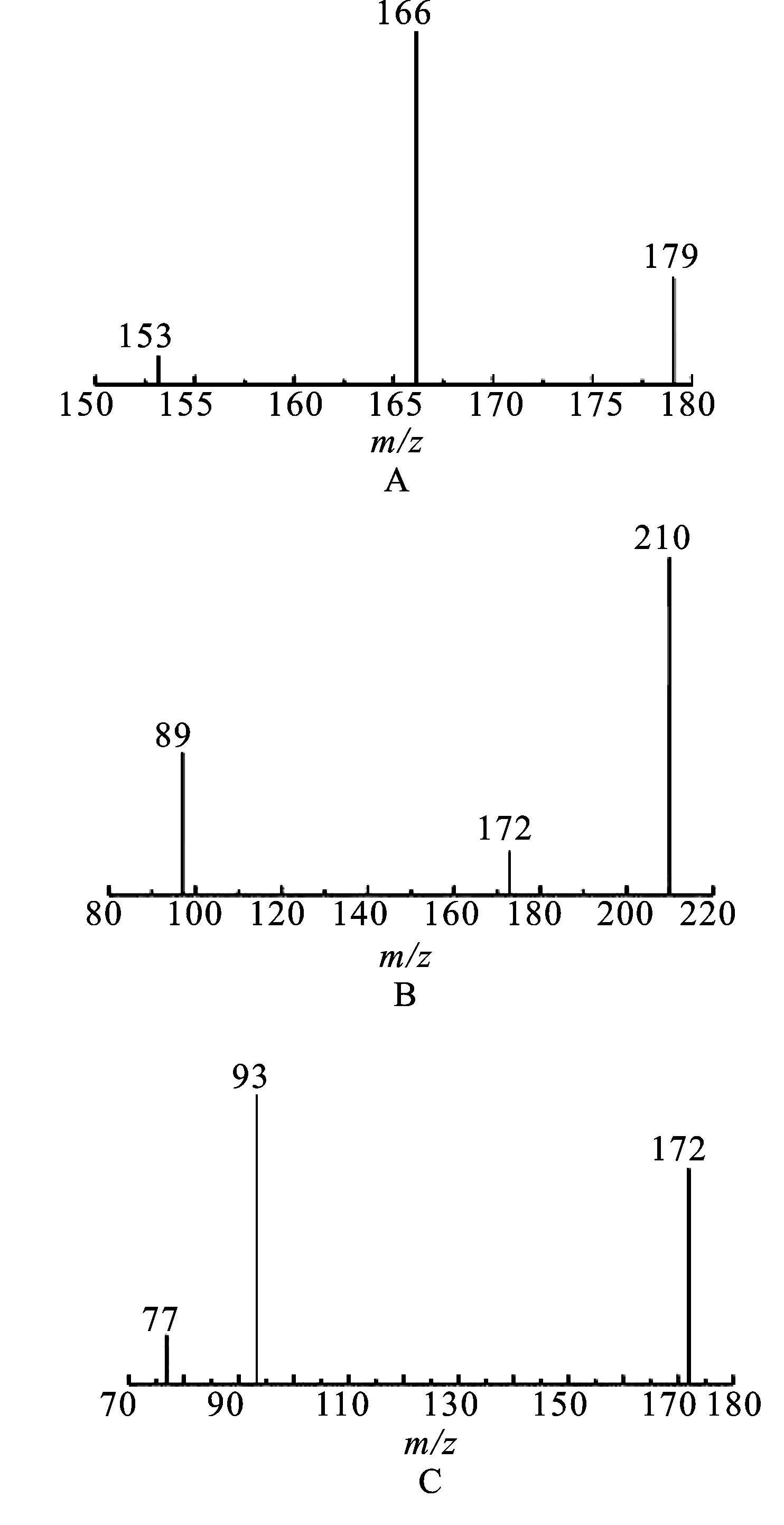
图2二级离子碎片图谱
A.联苯菊酯;B.甲氰菊酯;C.溴氰菊酯。
Fig.2 The chromatograms of secondary fragment ions
A.bifenthrin;B.fenpropathrin;C.deltamethrin.
2.6.2f值的耐受性考察 实际应用中的色谱条件可能会有一些变化,因此我们对可能影响f值的因素如对线速度和进样体积进行考察,结果表明,f值的波动较小,见表2。
表2相对校正因子耐受性考察
Tab.2 The tolerance investigation of relative correction factors
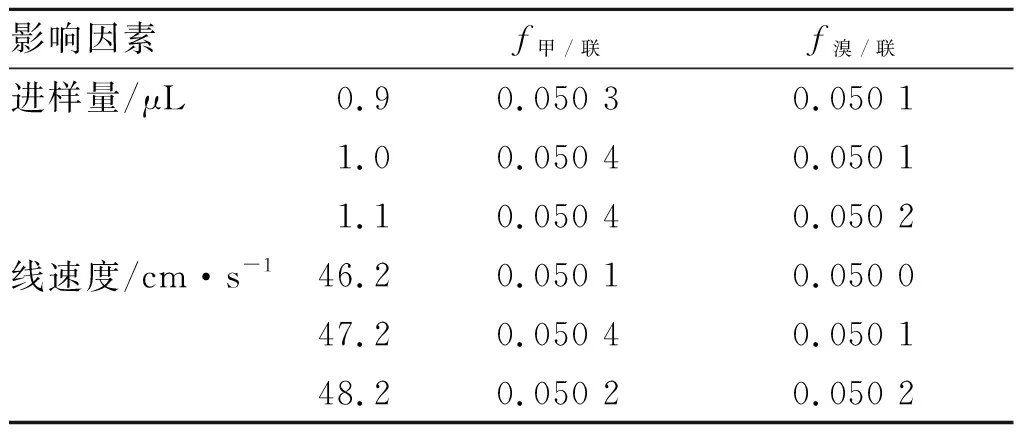
影响因素f甲/联f溴/联进样量/μL0.90.050 30.050 11.00.050 40.050 11.10.050 40.050 2线速度/cm·s-146.20.050 10.050 0 47.20.050 40.050 148.20.050 20.050 2
2.7精密度实验 精密量取2.2.1项下制备的对照品储备液20 μL,置于5 mL量瓶中,加丙酮稀释至刻度,摇匀。按照2.3项下测定条件重复进样6次,测得联苯菊酯二级离子扫描色谱峰(181.1/166.1)平均峰面积为411 233,RSD值为0.9%;甲氰菊酯二级离子扫描色谱峰(265.1/210.1)平均峰面积为20 918,RSD值为0.5%;溴氰菊酯二级离子扫描色谱峰(252.9/93.0)的平均峰面积为20 473,RSD值为0.7%。
2.8重复性实验 精密量取2.2.1项下制备的对照品储备液20 μL,分别置于6个5 mL量瓶中,加丙酮稀释至刻度,摇匀。按照2.3项下测定条件分别进样,测得联苯聚酯二级离子扫描色谱峰(181.1/166.1)平均峰面积为410 871,RSD值为0.3%;甲氰菊酯二级离子扫描色谱峰(265.1/210.1)平均峰面积为21 089,RSD值为0.8%;溴氰菊酯二级离子扫描色谱峰(252.9/93.0)平均峰面积为21 056,RSD值为0.4%。
2.9稳定性实验 取2.2.3项下制备的样品加标溶液,按照2.3项下条件分别在0,2,4,8,12,14,16,20和24 h进样,结果联苯菊酯、甲氰菊酯和溴氰菊酯二级离子扫描色谱峰面积变化较小,RSD值分别为1.0%,1.1%和0.8%。
2.10检出限与定量限 取2.2.3项下制备的样品加标溶液,按照2.3项下条件测定,测得联苯菊酯、甲氰菊酯和溴氰菊酯信噪比(S/N)分别为15.14,61.78和13.92,以S/N的3倍计算最低检出限、10倍计算最低定量限。结果联苯菊酯、甲氰菊酯和溴氰菊酯的最低检出限(以进样量计)分别为9.907×10-4,2.428×10-4和1.078×10-3ng;最低定量限(以进样量计)分别为3.303×10-3,8.093×10-4和3.592×10-3ng。
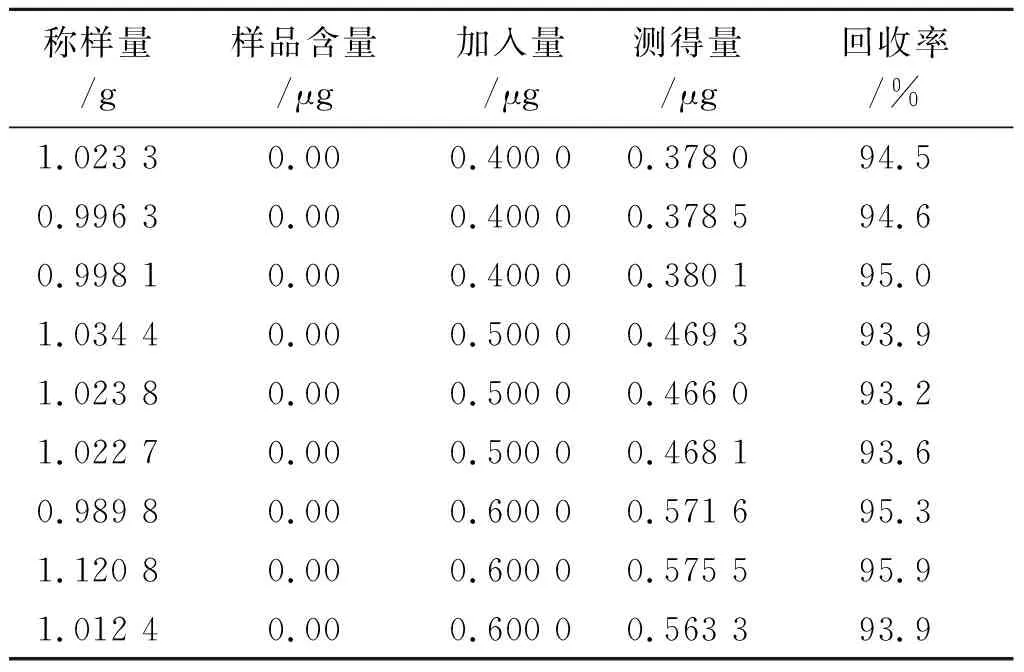
2.11回收率实验 取2.2.2项下粉末9份,分为3组,每组3份,每份1 g,精密称定,分别置于9个50 mL的离心管中,每组分别加入对照品储备液4,5和6 μL,按照2.2项下方法制备样品溶液,按照2.3项下测定条件分别进样,记录图谱,按照回归方程y=10 388 461x-236计算供试品中联苯菊酯的含量;按照校正方程A甲氰菊酯=10 388 461mf甲/联-236计算甲氰菊酯含量,按照校正方程A溴氰菊酯=10 388 461mf溴/联-236计算溴氰菊酯的含量。结果联苯菊酯、甲氰菊酯和溴氰菊酯的平均回收率分别为96.9%,97.2%和94.4%;RSD值分别为1.1%,1.0%和0.9%。结果见表3至表 5。
2.12样品测定 分别取批号为20170901,20170902和20170903的浙贝母样品50 g,粉碎,过三号筛,分别取粉末1 g,精密称定,按照2.2项下方法制备样品溶液,按照2.3项下测定条件分别进样,记录图谱,并按照2.10项下方法计算样品联苯菊酯、甲氰菊酯和溴氰菊酯残留量,结果表明,3批样品的残留量测定结果均为0 ng·kg-1。
表3联苯菊酯回收率实验结果
Tab.3 Results of the bifenthrin recovery test (n=9)
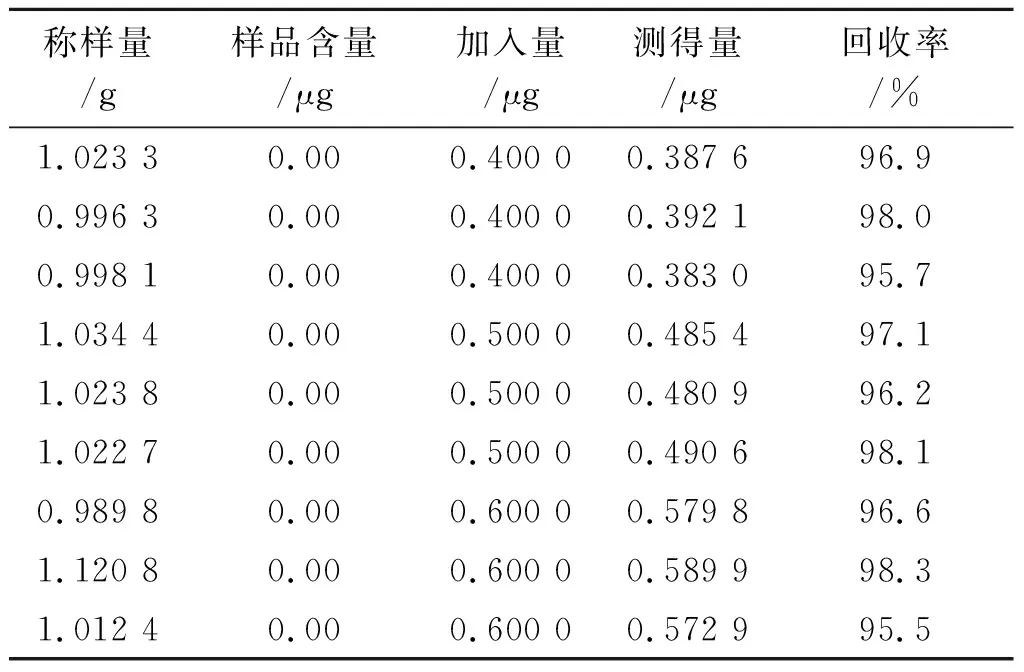
表4甲氰菊酯回收率实验结果
Tab.4 Results of the fenpropathrin recovery test (n=9)
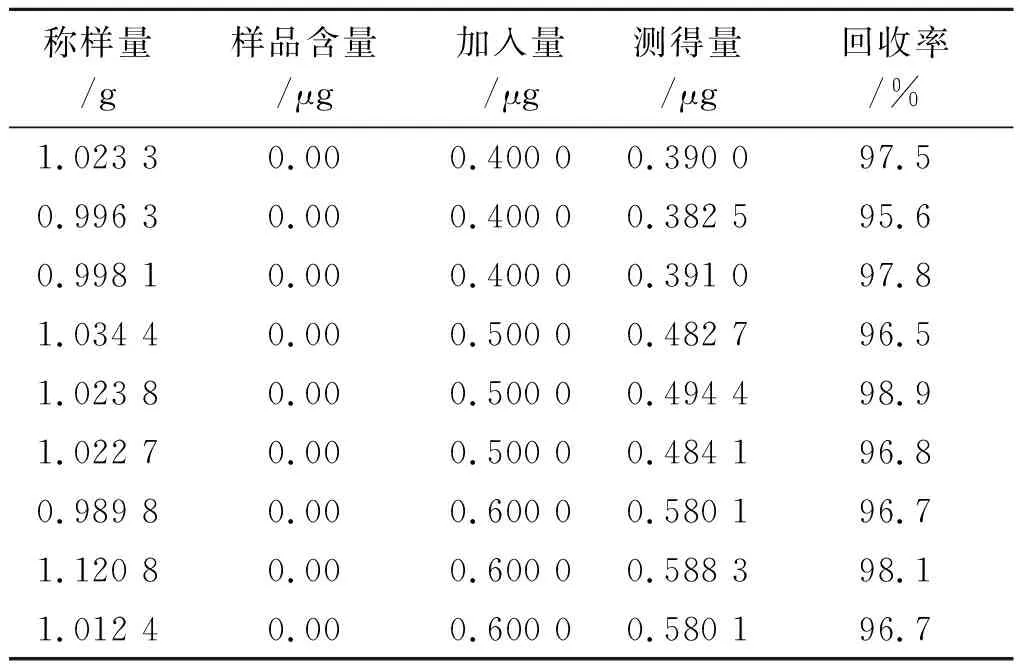
表5溴氰菊酯回收率实验结果
Tab.5 Results of the deltamethrin recovery test (n=9)
3 讨论
3.1耐受性考察 本实验中的相对保留时间可能会因色谱条件的细微改变而发生变化,但在该实验中,被测定物质的定性主要采用二级离子碎片峰及其丰度比确认,因此未对相对保留时间的耐受性进行考察。在对相对校正因子f值耐受性进行考察时,因仪器厂家建议进样体积不超过1 μL,因此对进样体积0.9,1.0和1.1 μL时的f值进行了比较,结果变化不大。
3.2校正因子的计算 校正因子计算法常应用于化学药有关物质的测定、中药材及其复方制剂中多指标成分的测定[17-19]。校正因子的表示方法很多,本实验的校正因子是指气相色谱法中的相对质量校正因子。校正因子的测定可精密取一个成分对照品和其他物质对照品各适量,分别配制成不同质量浓度的溶液进样,记录色谱图,绘制选定成分进样量和其他成分进样量对其峰面积的回归曲线,用回归直线斜率之间的比值来计算校正因子。
3.3提取溶剂的考察 选择提取溶剂时,因乙腈对联苯菊酯等农药溶解度较大,可溶入的杂质量适中,故选择乙腈为提取溶剂;在Cleanert TPH固相萃取柱上加入2 cm高的无水硫酸钠以除去样品溶液及洗脱液中的水份。
3.4GC-MS(MS/MS)联用法的优势 我们对多种中药材的农药残留含量测定进行了研究,由于中药材含有多种化学成分,会对测定有一些干扰,因此在测定具体某一品种时应设计具体的实验参数,并进行相应方法学论证。GC-MS(MS/MS)联用法既具有气相色谱的高分离效能,又具有质谱(二级质谱)可鉴定化合物结构的特点,可同时快速测定样品中多种残留农药及其衍生物。
3.5一测多评法的优势 一测多评法是以一种对照品来建立该成分与其他成分间的相对响应因子,通过相对响应因子来计算其他成分含量的质量控制方法,这是本文创新点,以往文献暂无此方法的详细报道。该法具有成本低、效率高等优点,能有效解决多指标质量控制面临的对照品短缺、不易获得、检测成本相对昂贵和农药对照品造成的环境污染等问题。
