范可尼贫血FANCA基因双重杂合突变1例报告并基因突变分析
2018-11-13杨娅平李栋梁曹玮赵田华
杨娅平,李栋梁,曹玮,赵田华
(1承德医学院,河北承德067000;2解放军白求恩国际和平医院)
范可尼贫血(FA)是一种罕见的常染色体或X染色体连锁的隐性遗传病,临床表现为进行性加重的造血功能衰竭、先天性畸形及高风险进展为急性髓系白血病[2],约50%的患者呈原发不孕[3,4]。FA的发生源于FA基因突变。FA基因是一组在DNA交联损伤中起同源重组修复(HRR)作用的基因[1],其常见的突变类型包括FANCA、FANCC和FANCG。目前认为,FA是由于DNA内切酶异常阻碍了受损DNA自然修复过程,进而引起染色体畸变,最终导致造血干细胞缺陷及多发性畸形。常规诊断FA主要依据全血细胞减少家族史、骨髓再生障碍、特征性先天畸形及染色体断裂等。目标序列捕获高通量测序是近年发展起来的一项新的测序技术,能够同时筛查多种基因突变。我们应用此技术检出1例FA患儿携带FANCA基因双重突变,用Sanger测序法加以验证并检测其父母、姐姐基因型。现报告如下。
1 资料与方法
1.1 临床资料 患儿女,9岁,因“间断乏力、鼻衄1年10月余”,于2015年11月6日来我院就诊。患儿既往在当地医院依据血常规、骨髓穿刺结果考虑再生障碍性贫血(具体检查结果未知),日常间断口服康力龙及环孢素治疗,疗效不满意。患儿父母非近亲结婚,家族中无类似疾病史。查体:发育落后于同龄儿,无骨骼畸形,中度贫血貌,皮肤未见色素沉着及出血点,心脏未闻及异常杂音,肝脾肋下未触及肿大。实验室检查:①血常规:白细胞3.0×109/L,中性粒细胞36.5%,淋巴细胞60.8%,红细胞2.6×1012/L,血红蛋白102.0 g/L,血小板38.0×109/L。②骨髓象:有核细胞增生减低,粒系比例略低,占26.5%,均为中幼及以下阶段,胞质颗粒明显增多;红系比例明显增高,占57.5%,以中晚幼红细胞为主,偶见双核红细胞,成熟红细胞大小不一;淋巴细胞比例基本正常;全片共见巨核细胞4个,血小板散在少见。③骨髓活检:骨髓增生极度低下(10%),伴明显出血,少量粒、红系细胞散在分布,分叶核为主,巨核细胞可见,偶见胞体小的巨核细胞。网状纤维染色(MF)0级。免疫组化:CD34、CD14、CD117阴性,CD61、CD42b、MPO阳性,CD68少数阳性。④骨髓增生异常综合征(MDS)及白血病相关基因检测:骨髓细胞FISH检查MDS、白血病相关基因EGR1、CEP7、TP53、CEP8均未见异常;Sanger测序示MDS、白血病相关基因IDH1、IDH2、NPM1、DNMT3A、SRSF2、SF3B1及U2AF1均未见异常。④染色体分析:核型为46,XX;染色体断裂实验未见异常。⑤肾脏B超:左肾缺如,右肾融合肾。考虑患儿年幼伴发先天畸形,发育迟缓,间断口服康力龙及环孢素症状缓解差,可能为骨髓衰竭综合征,尤以FA为著,因此做FA相关检查。以50例健康志愿者作为对照排除基因多态性。本研究经医院伦理委员会批准,并征得患儿及家属知情同意。
1.2 患儿FANCA突变基因检测及验证
1.2.1 基因组DNA提取 采集患儿外周血3~5 mL,EDTA抗凝,按DNA提取试剂盒说明书提取外周血DNA,-20 ℃保存备用。
1.2.2 FANCA突变基因检测 采用目标序列捕获和高通量测序技术。利用基于第二代测序方法设计的包含249种先天性骨髓衰竭症的诊断试剂盒,首先将提取的基因组DNA用超声波打断并制备DNA文库,然后通过捕获芯片对FANCA基因编码区及其侧翼序列的DNA进行富集,用通用引物对捕获到的序列进行PCR扩增,最后应用Illumina Hiseq测序平台对扩增产物进行高通量测序,寻找突变的致病基因。
1.2.3 FANCA突变基因鉴定 采用Sanger测序技术。在确定患儿突变的致病基因后,对患儿外周血DNA进行测序验证。引物设计采用Primer Premier5.0软件,由上海生工生物合成。PCR扩增反应体系(50 μL):DNA模板5 μL,10×PCR缓冲液5 μL,l×dNTP 5 μL,Taq酶2 U,上、下游引物各1.5 μL,补双蒸水至50 μL。反应条件:94 ℃ 30 s,55~65 ℃ 40 s,72 ℃ 45 s,共35个循环。将PCR产物在琼脂糖凝胶上进行电泳,紫外灯下观察。DNA测序:PCR产物纯化后,用测序试剂盒在ABI3130 DNA测序仪上进行序列测定。将检测结果与人类基因突变数据库(HGMD)进行比对,确定有无此类突变的报道。另外对50例健康人进行该基因突变的筛查作为阴性对照。
1.3 患儿家系直系成员FANCA突变基因检测 采集患儿父母、姐姐的外周血,采用Sanger测序技术检测外周血DNA的基因型。方法同上。
1.4 突变危害性的预测 应用Mutation Taster和PolyPhen-2软件预测基因突变对蛋白功能的影响程度。当两种软件均预测同一突变对蛋白的功能影响较大时,认定该突变具有较强的危害性。
2 结果
2.1 患儿FANCA突变基因检测结果 目标序列捕获测序分析FANCA基因上所有外显子的测序覆盖率均达到99%以上,各外显子上的平均测序深度和测序深度中位数均比较接近,提示测序的随机性比较好。通过目标序列捕获高通量测序技术检测患儿FANCA基因的所有外显子及其侧翼序列,发现16号染色体上FANCA基因编码区存在双重杂合突变,即第14外显子c.1303C>T(p.R435C)和第31外显子c.3031C>T(p.R1011C),导致该基因的第435位和1011位氨基酸均由精氨酸变成半胱氨酸。见图1、2。

图1 患儿FANCA基因c.1303C>T突变目标序列捕获和高通量测序图

图2 患儿FANCA基因c.3031C>T突变目标序列捕获和高通量测序图
2.2 患儿FANCA突变基因验证结果 应用Sanger测序法对先证者的上述突变位点进行验证,其结果与目标序列捕获高通量测序法完全一致。见图3、4。对50例健康个体FANCA基因相应区域测序,均未发现上述序列改变。
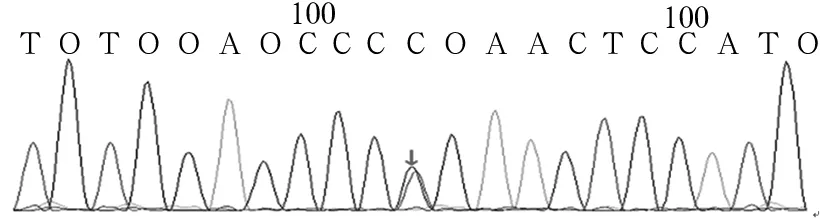
图3 患儿FANCA基因c.1303C>T突变Sanger测序图

图4 患儿FANCA基因c.3031C>T突变Sanger测序图
2.3 患儿家系直系成员FANCA突变基因检测结果 父亲携带第31外显子c.3031C>T(p.R1011C)突变,母亲及姐姐均携带第14外显子c.1303C>T(p.R435C)突变。
2.4 蛋白功能预测分析结果 c.3031C>T(p. R1011C)和c.1303C>T(p.R435C)对蛋白功能影响均为有害。
3 讨论
多数FA患者出生时血细胞正常或接近正常,在5~10岁时出现血细胞减少[5],常合并贫血、出血和感染,部分患者表现为进行性骨髓衰竭与继发性肿瘤如急性髓系白血病等,造血功能衰竭合并感染为该病常见的死亡原因。随着年龄增长,FA患者可出现多发畸形与发育迟滞,其中畸形的发生率约为60%[6]。FA的实验室检查包括血象、骨髓象及细胞遗传学检测等。其血液学改变程度和类型呈高度异质性;骨髓象类似于获得性再障,表现为增生减低、造血细胞减少、非造血细胞增多;细胞遗传学检测可见染色体断裂、缺失,染色单体互换、核内再复制、环形染色体畸变等不稳定现象。Auerbach等[7]报道了222例FA患者,其中44%表现为骨髓造血异常和先天畸形,51%仅有造血异常或先天畸形,5%两者皆无。但也有再障患儿伴先天畸形而无细胞遗传学改变者,因此仅靠临床表现可能漏诊或误诊。本例患儿自幼身材矮小,发育迟于同龄正常儿童,7岁左右出现乏力、鼻衄症状,血常规提示全血细胞减少,骨髓象提示骨髓造血功能低下,超声发现左肾缺如及右侧融合肾,符合FA患者发育迟缓、骨髓造血衰竭及先天畸形的临床特点。
近年来,利用体细胞融合杂交技术和FA细胞对DNA交链剂异常敏感的基因缺陷进行互补分析,目前已发现有19种基因被证实参与FA蛋白的相关功能。这些基因以“FANC”为词根进行命名,FANCA、FANCC和FANCG是3种较为常见的基因突变,约占FA患者的85%,其中FANCA基因亚型最为常见[8]。FANCA约有200个不同的等位基因,包括几乎所有的已知的突变类型,大片段的缺失是主要的突变类型。转化为骨髓增生异常综合征及白血病的FA通常有基因或染色体异常,其中7号和3q异常的预后差[9,10]。
目标序列捕获高通量测序是将目标序列捕获与高通量测序相整合进行DNA序列分析的新技术,具有速度快、准确率高、成本低、覆盖度广以及与生物学表型结合更为直接等优点,能够同时检测大量基因,适用于遗传异质性疾病的基因突变筛查,具有很好的应用潜力,亦为检测FA相关的基因突变提供了更为准确和直观的诊断依据。本研究应用该技术对1例疑似FA患儿进行了249种骨髓衰竭症相关基因突变的筛查,发现本例患儿存在16号染色体上FANCA基因第14外显子c.1303C>T和第31外显子c.3031C>T双重杂合突变,并用Sanger测序得以验证,从而确定了FA的诊断。有关FANCA基因14外显子c.1303C>T突变国内外仅见1例报道,即在97例FA患者中检测到40种致病基因突变位点,其中1例为c.1303C>T突变,且此突变与FA发病密切相关[11];后一突变位点通过查询千人基因组计划数据库(HGMD)和美国国家生物技术信息中心单核苷酸多态性数据库(dbSNP)目前尚未见报道。我们进一步应用蛋白功能预测软件检索提示,上述2个突变对蛋白功能影响均为有害。
FA的治疗首选异基因造血干细胞移植,可修复受损细胞及改善FA造血微环境,是提高患者长期无病生存率的惟一方法;无移植条件者可给予雄激素、细胞因子如粒细胞集落刺激因子及对症支持治疗。该患儿平时间断口服雄激素及输血治疗,目前病情稳定,正在进一步随访中。
