中国汉族常染色体显性遗传颞叶外侧癫痫家系的临床特征和基因突变筛查
2018-10-30李承玉徐连萍杨华俊王群
李承玉徐连萍杨华俊王群△※
1995年由OTTMAN等[1]首先报告的一种以听觉症状为特征的家族性部分性癫痫,即常染色体显性遗传颞叶外侧癫痫(autosomal dominant lateral temporal lobe epilepsy,ADLTE),又称为伴听觉特征的常染色体显性遗传部分性癫痫(autosomal dominant partial epilepsy with auditory features,ADPEAF),是一种罕见的特发性部分性癫痫综合征,患者表现为伴有听觉先兆或其他感觉性先兆的局灶性发作,可继发全面强直-阵挛性发作,多数药物治疗有效。2002年KALACHIKOV等[2]首次发现LGI1是其致病基因,后续在不同人群的研究中陆续得到证实,2015年DAZZO等[3]又发现一些ADLTE家系存在RELN基因突变。目前亚洲裔ADLTE患者的临床和相关致病基因研究较少,我们对4个中国北方汉族裔ADLTE家系的临床表现及基因筛查结果进行总结和分析,并综合既往文献进行探讨。
1 对象与方法
1.1 研究对象和临床资料采集4个家系的先证者为2016年5月-2018年1月在北京天坛医院癫痫科就诊的患者,均为中国北方汉族裔,由两位经验丰富的癫痫专科医师作出ADLTE的临床诊断,诊断标准包括:①表现为具有听觉特征的局灶起源的癫痫;②头颅MRI等影像学未发现病因性异常,且神经系统查体为阴性;③家族中有≥2例癫痫患者。所有患者均有头颅MRI检查和至少一次的发作间期脑电图检查结果,通过病史追溯、影像学检查等排除继发性癫痫。在取得患者的书面知情同意后,由1位经过遗传学训练的医师采用面访和电话访视的方式进行遗传背景调查,绘制家系图,收集患者的临床信息、影像学和脑电图等资料。
1.2 基因组DNA提取和遗传学分析采集患者的外周静脉血5 mL,通过标准方法提取DNA。采用美国Agilent公司含2742个人类已知致病基因的检测试剂盒,对先证者进行目标基因靶向测序,采用测序平台Illumina Hiseq X Ten(赛福基因,北京),参考基因组hg19,重点分析LGI1和RELN基因。数据与遗传学开放数据库1000 genome,db-SNP和ExAC进行比对,并以854个中国健康人的全外显子测序(whole exon sequence,WES)数据(北京,康旭基因)作为同种族裔对照。对上述所发现的变异使用 Mutation Taster (http://www.mutationtaster.org) 、SIFT (http://sift.bi.a-star.edu.sg/)和 PolyPhen2(http://genetics.bwh.harvard.edu/pph2/)进行功能预测。对所发现的可能致病的基因变异,根据既往文献提供的方法进行直接PCR扩增并Sanger测序验证[3-4]。
1.3 随访所有患者随访至2018年6月30日,随访资料来自门诊访视或电话随访。
2 结果
2.1 基本临床资料我们共收集到来自4个家系的ADLTE患者17例,发病年龄为12~37岁,多数在 10~20 岁之间起病(64.7%,11/17),男女性别比约为1.43(10∶7)。家系L三代中有2例癫痫患者(图 1a)。先证者 III2(男,22 岁)16 岁起病,第一种发作为右侧耳鸣,伴紧张、恐惧感,随后意识丧失、肢体抽搐,数分钟缓解,此种发作仅有3~4次;另一种发作仅有右侧耳鸣伴恐惧感,每次持续数秒至十余秒,每个月有2~3次,在服用左乙拉西坦0.75 g每天2次后第一种发作完全控制,但在熬夜、情绪不佳时偶出现第二种发作;患者的间期脑电图为双侧额颞区尖波,头MRI未见明显异常。II5为先证者的叔叔,自15岁起病,发作与其相似,表现为右侧耳鸣,意识清楚,十余秒好转,发作的间隔不定,平均每年4~5次,期间有3~4次在耳鸣后出现意识不清伴肢体抽搐的发作 (继发全面强直阵挛性发作,sGTCS),服用苯巴比妥溴化钠治疗后患者已10余年无发作;患者II5的发作间期脑电图和头颅MRI均未见异常。
家系Y五代共有癫痫患者8例(图1b),先证者为III4,家系中有3例患者已故,死因均非神经系统疾病。先证者为男性,57岁,15岁首次发作,感觉周围出现嘈杂、高调噪音,随后意识不清、牙关紧咬、四肢抽搐(sGTCS),每次约 3 min,最初数年不发作,从5年前起每年1~3次;有时仅有听觉症状,持续十余秒后缓解,此种发作较为频繁,每周均有十余次;患者的头颅MRI和间期脑电图未见明显异常;服用奥卡西平后目前3年无发作。先证者的同代有4例癫痫患者,其中III6(男,49岁)17岁初次发作,表现为睡眠中突然醒来,随后双侧耳鸣,声音逐渐变大,约10 s后意识丧失、双眼上翻并伴面部及四肢抽搐,3~5 min钟后缓解,最初每年发作1~2次,服用中成药后25岁~43岁之间无发作,5年前再发,间断服用卡马西平,服药期间可控制无发作。患者IV1(女,30岁)自22岁生育后开始发作,发作时感周围声音嘈杂,同时无法说话但能听懂他人言语,数秒钟后意识丧失并四肢抽搐,患者服用拉莫三嗪3年后发作完全控制,目前已超过5年无发作。家系中患者III2(女,65岁)和 III9(女,51岁)分别在 19岁、17岁左右出现首次发作,表现为sGTCS,无明显听觉或视觉症状,多数在夜间出现,其中III2仅有4次睡眠中的发作,而III9发作较频繁,但在服用卡马西平和丙戊酸镁治疗后可控制。除先证者III4的发作间期脑电图可见右侧前中颞区尖慢波外,其余患者的发作间期脑电图均未见明显异常;所有患者头颅MRI均未见异常。
家系Z中共4例患者 (图 1c),先证者 III2(男,17岁)从12岁时开始发作,表现为电话铃声刺激诱发的意识不清伴四肢抽搐,每次持续约2~3 min,病程最初每半个月1次,服用奥卡西平0.45 g每天2次后发作完全控制,患者的发作期脑电图显示为双侧颞区非同步性尖波,头颅MRI提示小脑蛛网膜囊肿。患者II7(男,29岁)的发作期症状为听到声音逐渐变小,数秒钟后出现意识不清、四肢抽搐;脑电图上有右侧颞区尖慢波。患者I2(女,71岁)和 II2(女,40岁)的听觉特征不明显,表现为意识丧失伴四肢抽搐(sGTCS)。 I2、II2、II7的间期脑电图和头颅影像学均未见明显异常。
家系D (图2)共3例癫痫患者,III4为先证者,14岁首次发作,发作期症状有典型的听觉特征 (先听见模糊说话声或右耳听见高调性耳鸣声),这种发作大约每天3~4次,同时还有间隔数月1次的sGTCS发作,奥卡西平加量至0.6 g每天2次后未再发作。患者II5为先证者的母亲,自17岁开始发作,发作有时由高调的电话铃声诱发,发作时表现为sGTCS,每1~2年有一串季节性发作,服用丙戊酸钠治疗后近7年无发作。患者III3为先证者的同卵双胞胎哥哥,12岁起病,但发作期无明显听觉、视觉或失语等症状,每年仅有1~2次的sGTCS,服用丙戊酸钠缓释片500 mg每天2次后仍偶有发作,但发作均与停药、漏服药相关。本家系中3个患者的间期脑电图、头颅MRI均未见异常。上述4个家系的临床特征总结见表1。
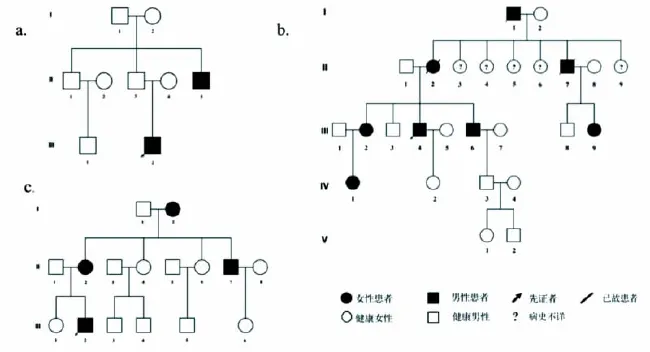
图1 家系L、家系Y和家系Z的遗传图谱
2.2 遗传检测及生物信息学分析结果通过上述遗传学检测和分析方法,我们发现家系D的3例患者的LGI1基因具有共同的杂合性c.367G>A(p.E123K)变异(图 2b),Sanger测序在家系中进一步验证了上述变异的存在,且家系中疾病表型和基因型共分离(co-segregation)。该变异位点在1000Genome、ExAC、dbSNP 等健康人数据库中未出现,且在854名中国健康人的WES样本数据库中未检出,说明该变异在健康人中不携带。进一步生物信息学分析发现,该变异位于蛋白的第3个LRR结构域,SIFT预测在物种进化中相对保守,为对蛋白结构有害的(deleterious),且在Polyphen-2中提示很可能致病性(probably damaging)。除此以外,我们还在患者中发现了1种基因错义变异(c.1219C>T)和 6 种基因错义变异(c.1521A>G、c.1690C>A、c.8863C>T、c.1570T>C、c.3941G>A),均为杂合性,上述变异经过与健康人数据库比对后证实均属于单核苷酸多态性(SNP)。本研究发现的SNP见表2。
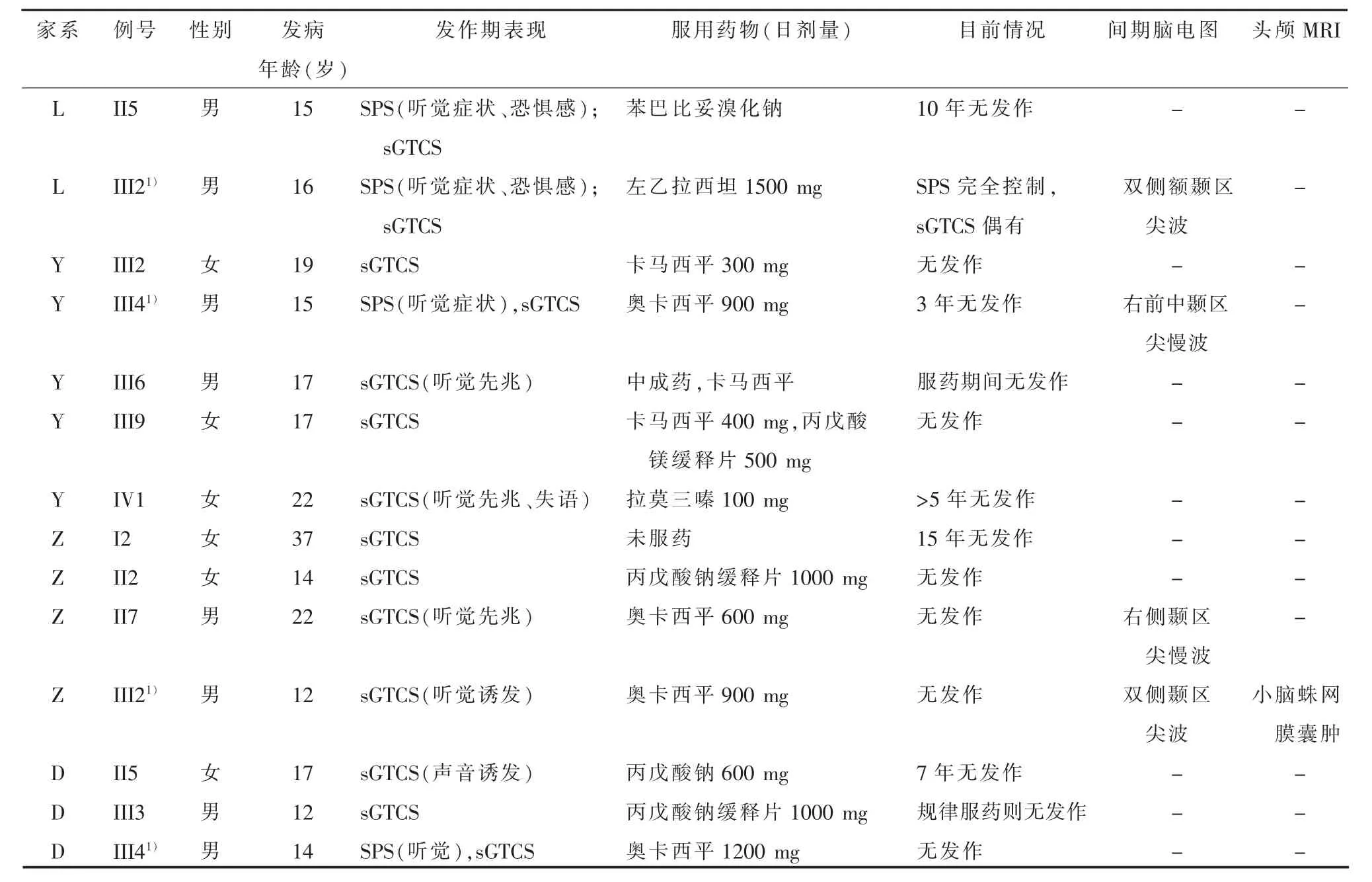
表1 4个家系所有存活患者的临床特征
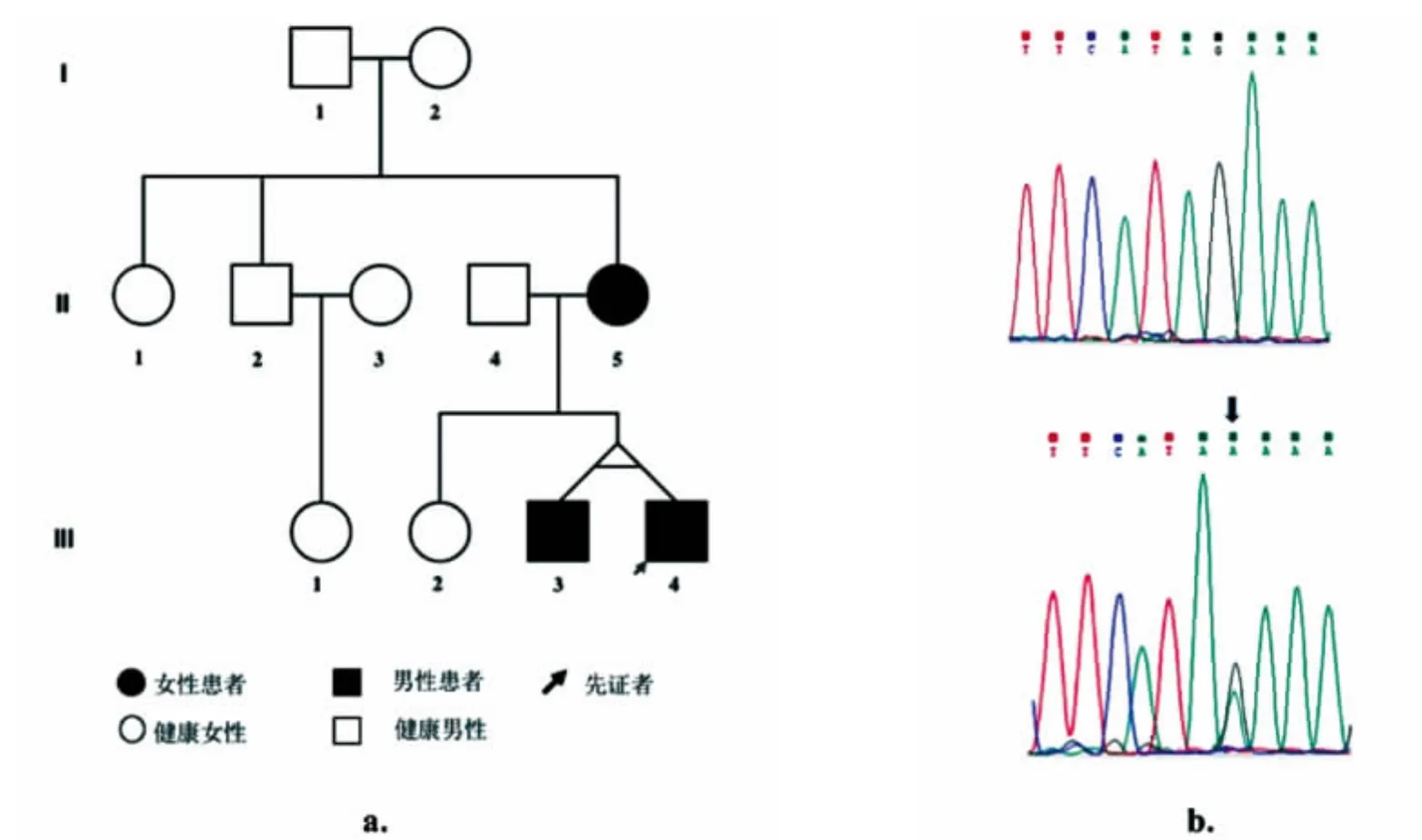
图2 (a)家系D的遗传图谱;(b)家系中3名患者均携带LGI1基因杂合性错义变异c.367G>A(箭头所示),而家系中未患病成员II4和III2该位点未见变异
3 讨论
ADLTE是一种罕见的癫痫综合征,多数为家族性,也有少数散发病例报告[5]。患者的首次癫痫发作时间多在青春期或成人早期,男女患病比例大致相同,多数患者发作期具有典型的听觉症状,多为简单声音幻觉,有时可以有复杂的声幻觉或眩晕,一部分患者还可以有视觉症状、失语等症状,一些患者的发作能被听觉刺激所诱发,这些都提示发作可能起源于颞叶外侧或与其相关联的新皮层[2,5-7],但研究发现约10%患者也可能出现颞叶内侧的症状,如似曾相识感(déjà-vu)、恐惧、发作期的精神症状甚至典型的复杂部分性发作。目前倾向ADLTE为一种良性的癫痫综合征,对传统或新型抗癫痫药均控制有效,但停药也可能容易复发[8]。本组报道的病例基本符合ADLTE的特点,发病多在青春期,但家系Y的患者较平均起病年龄稍晚;超过50%的患者发作期症状具有典型的听觉特征,提示放电起源于颞叶外侧面,发作期失语见于1例患者;除一名患者发现小脑蛛网膜囊肿外其余患者的头颅影像学均未见结构性异常,本组患者的上述临床特点与文献报告的典型ADLTE基本一致。
ADLTE具有一定的遗传异质性,LGI1是最常见的致病基因,所编码的LGI1蛋白为非离子通道蛋白,通过在神经元突触后膜与其配体神经元特异性膜蛋白ADAM22结合形成蛋白复合物,参与谷氨酸-AMPA神经传递;也可通过参与电压门控钾离子通道亚单位Kv1.1相关蛋白复合体的组成,发挥选择性阻碍Kvbeta1中介的N-型钙离子通道的激活,LGI1基因的突变可能导致离子通道快速失活功能丧失,从而使癫痫易感性增加,这可能是LGI1基因突变引起癫痫发作的机制之一。ADLTE患者的LGI1基因多为点突变,但也有报道存在基因片段缺失或基因微重排[6,9];引起典型ADLTE症状的突变多位于LRR结构域(第3~5号外显子)[10]。ADLTE患者LGI1突变报道最多的为白种人,但也仅占家系患者的不足50%[2],散发患者的阳性率更低,这表明ADLTE是一种遗传异质性的疾病,而这种异质性在非白人裔人群中则更明显——目前亚洲患者中仅日本、韩国有少数LGI1基因阳性家系个案报道,本课题组也在近期报道了中国首个LGI1基因突变的家系[11],但总体而言亚裔患者中LGI1基因检出率很低,这提示亚洲裔患者存在其他致病基因的可能性[12-13],但目前为止尚未有亚洲患者其他致病基因的相关报道。
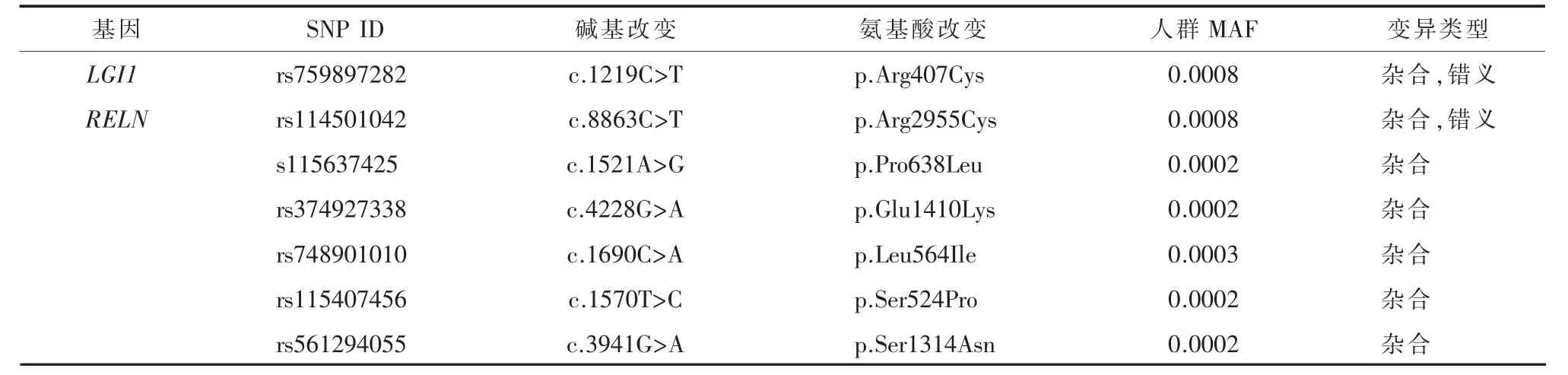
表2本研究中发现的LGI1和RELN基因SNP
RELN基因是目前为止发现的第二个ADLTE的致病基因。2015年DAZZO等[3]对40个LGI1基因阴性的意大利ADLTE家系进行SNP-阵列连锁分析和WES分析,在3个家系中发现了与疾病共分离的RELN基因错义变异,再对其他家系进行验证又发现了另外4个RELN基因的错义突变,最终RELN基因错义突变见于该研究中约17.5%(7/40)的家系。发生错义变异的位点多位于Reelin重复序列的结构域,后者为350~390个氨基酸残基组成的串联重复结构,包含一个位于中心的表皮生长因子(EGF)或EGF样模块(EGF-like module)及侧翼的两个子重复序列,三者共同构成Reelin重复序列的马蹄样结构,与Reelin蛋白的功能密切相关。变异引起上述Reelin重复序列的空间结构异常,从而可能使肝细胞对Reelin蛋白的外分泌减少,引起患者血清中和脑细胞外Reelin蛋白含量的降低[3]。后续研究又对这7个RELN基因突变的ADLTE家系的28名患者进行了临床表型分析,发现患者发病的平均年龄为20岁,约71%患者出现局灶性发作,其中约三分之一有失语、视觉异常和其他不常见症状(眩晕或似曾相识感),约8%的患者的发作可有环境噪音诱发,且绝大多数(96%)患者接受AEDs治疗可以获得完全无发作或发作减少50%,80%的患者的间期脑电图出现颞区的痫性放电,头颅MRI无显著异常。总体上RELN突变的患者临床表型与LGI1突变的患者表型无显著差异。
本项研究在4个中国汉族裔ADLTE家系中对上述已知致病基因的进行筛查,最终在家系D中发现LGI1基因4号外显子的一个杂合性错义变异c.367G>A(p.E123K)。该变异在生物信息学分析中为对蛋白结构有害,且Sanger测序验证了在家系中存在共分离现象。2009年DI BONAVENTURA等[14]在意大利ADLTE家系患者中曾发现过该突变,在不同种族背景的多个家系中该突变与疾病共分离,根据ACMG遗传变异分类标准与指南可作为其强致病性的证据。该意大利家系表现为听觉症状突出的癫痫发作,但携带突变基因的患者对多种AEDs耐药,与本研究中家系D的临床特征有所不同。本研究对RELN基因进行了检测,发现RELN基因6个错义变异,但经生物信息学分析,6个错义变异均不具有致病性。因此目前还没有找到证据表明RELN基因与中国ADLTE相关,这可能需要后续更多的家系和病例研究提供进一步的依据。同时本研究中除家系D外的3个ADLTE家系中亦未发现其他已知的与癫痫相关的致病基因突变,这也提示对于中国裔的ADLTE这种独特的遗传性癫痫综合征,可能还存在某些目前尚且未知的致病基因,值得我们进一步研究和探索。
