含乙烯功能团的β-环糊精单体的合成与表征
2018-10-24朱玭玭凌辉博张国权
朱玭玭,韩 笑,江 昊,凌辉博,张国权
(西北农林科技大学食品科学与工程学院,陕西杨凌 712100)
环糊精聚合物(CDP)是含有多个环糊精单元的高分子衍生物,既保持了环糊精自身包合、缓释和识别的能力,又兼具高聚物良好的机械强度、稳定性和化学可调性等,因而广泛应用于化学分离[1-4]、环境保护[5]、医药[6-7]等方面。除此之外,环糊精聚合物在食品领域的应用日益广泛[8-13]。
环糊精聚合物可由乙烯基或烯丙基环糊精直接聚合而得,而含乙烯基环糊精单体的合成,尤其是单取代乙烯基单体的合成,比较有代表性的是Harada等[14]采用丙烯酸间硝基苯酯与环糊精,在碱性水溶液中,通过酯交换法制得单-2-丙烯酸环糊精酯单体,但该法产率低(11%),后续处理麻烦。国内西工大范晓东课题组在制备可聚合环糊精单体方面做了大量工作。黄怡等[15-18]采用1,3-二环己基碳化二亚胺缩合法(DCC)、丙烯酸间硝基苯酯酯交换法和丙烯酰氯直接酰化法,制备分子中具有单烯丙基修饰和多烯丙基修饰的β-环糊精(β-CD)。三者相比,DCC缩合法具有实验条件温和、产物易分离提纯、且产率高的优点,但仍存在原料反应不彻底和易生成副产物等缺点。季文等[19]以NaOH为催化剂,利用3-溴丙烯得到分子中含有双键的环糊精衍生物,虽然实验操作简单,但耗时久,最佳反应时长需48 h。此外,朱久进等[20]利用单6-乙二胺β-环糊精与马来酸酐,在相对温和的条件下反应,得到含双键和羧基的双功能基环糊精单体,该单体既可与其它单体聚合得到新型环糊精聚合物,也可与其它聚合物反应得环糊精高聚物。由此可见,研究并寻找新的合成方法与技术具有十分重要的意义。
为了提高可聚合环糊精单体的制备效率并增加其种类,本文采用化学修饰的方法,在大环骨架基本不变的情况下,通过酰胺化反应和亲核反应在β-CD分子中引入双键,合成三种含有乙烯基或烯丙基的环糊精单体,并利用质谱、红外光谱以及核磁共振氢谱对三种单体的结构进行表征。
1 材料与方法
1.1 材料与仪器
β-CD(分析纯,98%,使用前重结晶2次,90 ℃真空干燥24 h)、对甲苯磺酰氯(p-TsCl,分析纯,99%)、丙烯酸(AAc,分析纯,98%)、N,N-二甲基甲酰胺(DMF,分析纯,99.8%)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI,分析纯,99%)、1-羟基苯并三唑(HOBT,分析纯,99%)、N,N-二异丙基乙胺(DIPEA,分析纯) 上海萨恩化学技术有限公司;丁香酸(SA,分析纯,99%)、丙烯酰氯(分析纯,98%)、3-溴丙烯(分析纯,98%) 上海维塔化学试剂有限公司;盐酸、丙酮、甲醇、无水乙醇等试剂 均为分析纯,新三力化玻站。
Vetex-70傅立叶变换红外光谱仪 德国布鲁克公司;AVANCE-Ⅲ核磁共振仪 瑞士布鲁克公司;LCQ Fleet电离质谱仪 赛默飞世尔科技(中国)有限公司;DHG-9123A电热恒温鼓风干燥箱 上海精宏实验设备有限公司;真空干燥箱 上海福玛实验设备有限公司;R-100旋转蒸发器 瑞士步琦公司。
1.2 实验方法
针对环糊精分子结构上特点,将环糊精分子上羟基转化为高反应活性胺基,再分别与丙烯酸、丙烯酰化丁香酸通过酰胺反应,制备6-丙烯酰乙二胺-β-环糊精(β-CD-6-EA)、6-丙烯酰化丁香酸-β-环糊精(β-CD-6-SA-AC)。另外,在NaH催化作用下,采用较温和的反应条件合成6-O-烯丙基-β-环糊精(Allyl-β-CD)。具体合成路线如图1所示。
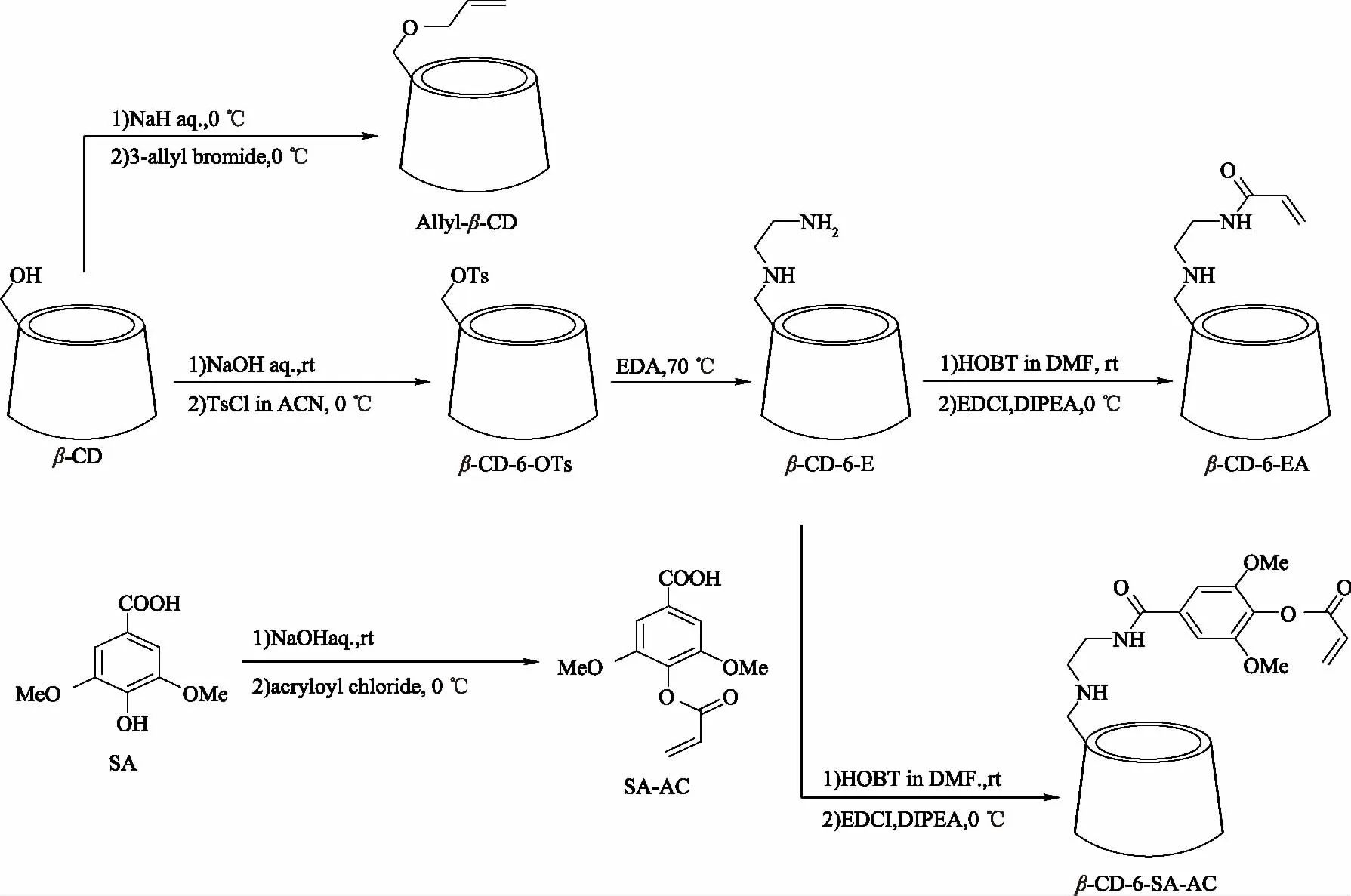
图1 含乙烯功能团的β-环糊精单体的合成路线Fig.1 Scheme representation for the synthesis of β-CDmonomers with vinyl functional groups
1.2.1 单6-对甲基苯磺酰-β-环糊精酯(β-CD-6-OTs)的合成 3.0 gβ-CD分散在25 mL蒸馏水中,0.34 g NaOH溶解在1.0 mL蒸馏水中边搅边滴到β-CD悬浮液中,再将0.75 g p-TsCl溶解1.5 mL乙腈中,冰浴条件下缓慢滴至上述悬浮液中,25 ℃搅拌反应2 h。用稀盐酸中和至pH=6.0,抽滤得粗产物[21]。粗产物在85 ℃热水中重结晶2次,60 ℃,0.088 MPa真空干燥24 h,得到白色固体产物0.53 g(产率:16%)。
1.2.2 单6-乙二胺β-环糊精(β-CD-6-E)的合成 3.0 gβ-CD-6-OTs与20 mL乙二胺混匀后,70 ℃反应4 h。之后,65 ℃旋蒸除去过量的乙二胺,再将余留物分散在400 mL丙酮中,抽滤得到粗产物。粗产物先后用3.0~4.0 mL蒸馏水、蒸馏水/甲醇(1∶3)混合液溶解,再逐滴加入400 mL丙酮中使产物析出,60 ℃,0.088 MPa真空干燥24 h得白色固体产物2.8 g(产率:99%)。
1.2.3 丙烯酰化丁香酸(SA-AC)的合成 冰浴条件下,将3.0 g丁香酸加到NaOH溶液(0.030 mol/L,40 mL)中搅拌。体系澄清后,逐滴加入1.5 mL丙烯酰氯。氮气保护,冰浴反应1 h,室温反应1 h。之后用稀盐酸中和析出固体[22]。粗产物先用45 ℃温水洗涤,再用45 ℃无水乙醇溶解,趁热过滤除去杂质,滤液冷却至室温放入4 ℃冰箱过夜。将析出物过滤后,40 ℃,0.088 MPa真空干燥24 h得到白色固体产物2.6 g(产率:70%)。
1.2.4 单6-丙烯酰乙二胺-β-环糊精(β-CD-6-EA)的合成 称取1.8 gβ-CD-6-E和0.31 g HOBT溶于20 mL无水DMF中。体系澄清后,冰浴下依次加入140 μL AAc、835 μL DIPEA和0.44 g EDCI,反应12 h。将反应液分散在350 mL丙酮中,抽滤得粗产物。用2.0~3.0 mL蒸馏水将固体溶解,再逐滴加入350 mL丙酮中反复纯化2~3次后,40 ℃,0.088 MPa真空干燥12 h得到淡黄色固体产物1.6 g(产率:88%)。
1.2.5 单6-丙烯酰化丁香酸-β-环糊精(β-CD-6-SA-AC)的合成 称取1.8 gβ-CD-6-E、0.46 g SA-AC和0.31 g HOBT溶于20 mL无水DMF中。体系澄清后,冰浴下依次加入835 μL DIPEA和0.44 g EDCI,反应12 h。将反应液分散在350 mL丙酮中,抽滤得粗产物。后续纯化、干燥方法同1.2.4,最终得到淡黄色固体产物1.7 g(产率:80%)。
1.2.6 6-O-烯丙基-β-环糊精(Allyl-β-CD)的合成 称取2.3 gβ-CD溶于30 mL无水DMF中,冰浴下加入0.25 g NaH搅拌1 h,再逐滴加入500 μL3-溴丙烯,室温反应7 h。将反应液分散在400 mL丙酮中,抽滤得粗产物。后续纯化、干燥方法同1.2.4,最终得到白色固体产物2.1 g(产率:90%)。
1.2.7 表征方法
1.2.7.1 质谱 采用电离质谱仪进行结构表征,室温下测定,分子量扫描范围80~2000 m/z。样品制备:取10 mg左右的样品,滴加DMF使其完全溶解即可。
1.2.7.2 傅立叶红外光谱 采用傅立叶变换红外光谱仪扫描各单体的FT-IR谱图。样品制备(KBr压片法):称取1 mg样品和100 mg溴化钾置于玛瑙研钵中,充分研磨混匀,然后移置于压模中进行压片。扫描范围为4000~400 cm-1,分辨率为4 cm-1,扫描次数为16次。
1.2.7.3 核磁共振氢谱(1H-NMR) 采用核磁共振仪进行结构表征。样品制备:取20 mg左右的样品,用500~600 μL D2O溶解,再转移至核磁管即可。
2 结果与分析
2.1 SA-AC的结构表征
由SA-AC的质谱结果如图2A所示:基峰对应的质荷比250.99是由[SA-AC(252.06)-H(1)]+形成的,表明丙烯酰化丁香酸成功合成。
由SA-AC的红外图谱图2B可知:位于3373.01 cm-1处丁香酸中-OH的伸缩振动吸收峰消失,而1752.72 cm-1处出现C=O的伸缩震动吸收峰。由此可见,丁香酸分子中的羟基与丙烯酰氯成功发生酰化反应。
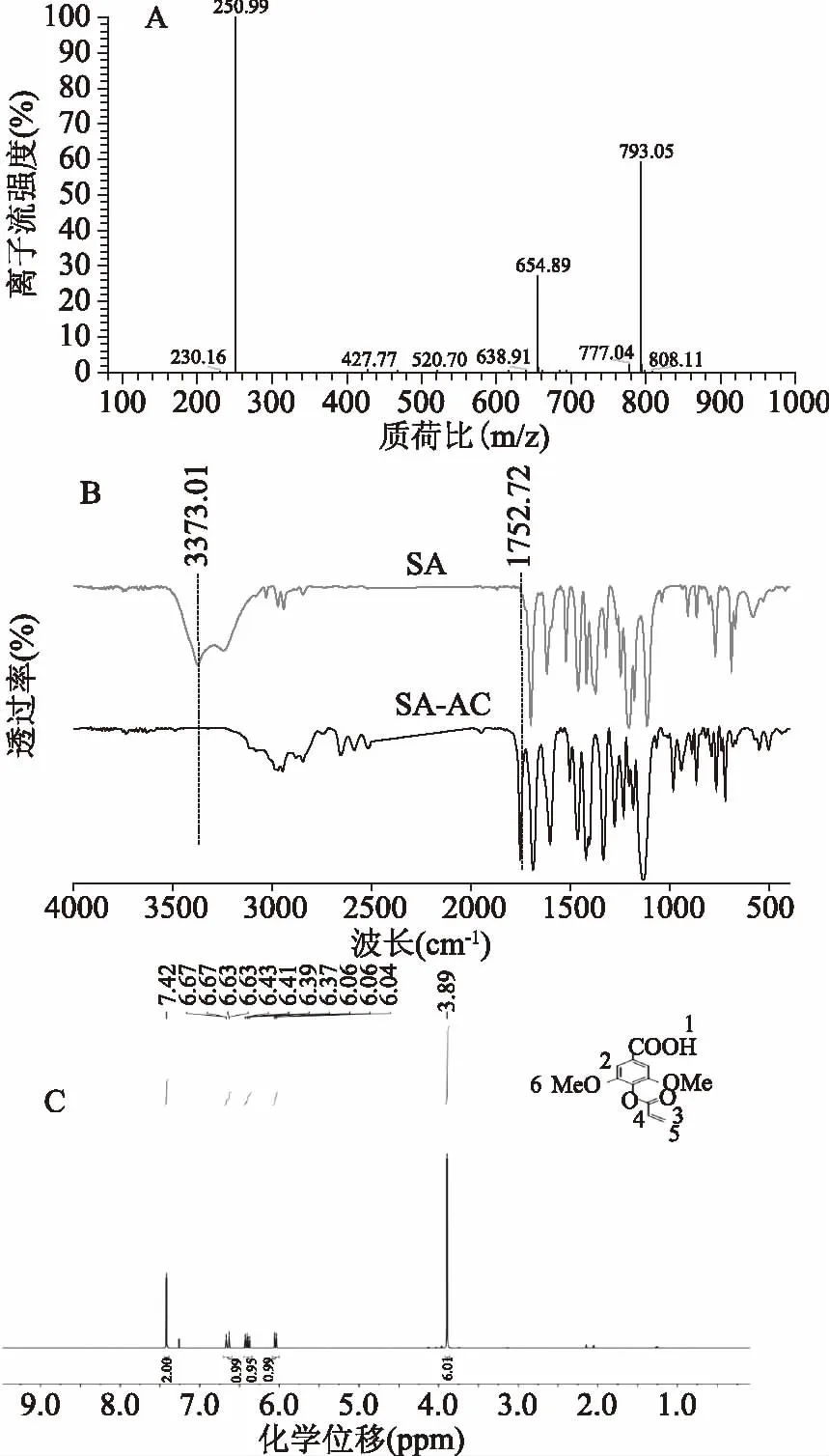
图2 SA-AC的质谱图(A)、傅立叶红外谱图(B)和核磁共振氢谱图(C)Fig.2 Mass,FTIR and 1H-NMR spectrum of SA-AC
采用1H-NMR对SA-AC的结构进行表征,由2C可知:(500 Hz,D2O)δ 7.42(s,2H,Ar),δ 6.67~6.63(dd,1H,J=1,17.5 Hz,Alkene),δ 6.4~6.37(dd,1H,J=10.5,17.5 Hz,Alkene),δ 6.06~6.04(dd,1H,J=1,15 Hz,Alkene),δ 3.89(s,6H,-OCH3)。其中,7.42和3.89分别是苯环单元上C-H和-OCH3的质子峰且都为单峰;6.67~6.04是丙烯基中氢的质子峰。由此可见,目标产物SA-AC已成功合成。
2.2 β-CD-6-EA的结构表征
β-CD-6-EA的质谱结果如图3A所示:基峰对应的质荷比1231.52是由[β-CD-6-EA(1230.44)+H(1)]+形成的,表明终产物以单取代为主。
由β-CD-6-EA的红外图谱图3B可知:位于1365.50 cm-1和1178.21 cm-1处β-CD-6-OTs的特征吸收谱带均消失,表明化合物已由β-CD-6-OTs转化为β-CD-6-E;1656.01 cm-1(仲酰胺Ⅰ谱带)是由酰胺的C=O伸缩振动产生的,1556.99 cm-1(仲酰胺Ⅱ谱带)是由N-H弯曲振动和C-N伸缩振动组合吸收产生的,1636.19 cm-1为C=C伸缩振动吸收峰[15]。由此可见,β-CD-6-EA已由β-CD-6-E酰化得到。
采用1H-NMR对β-CD-6-EA的结构进行表征,由图3C可知:(500 Hz,D2O)δ 7.41~7.38(m,1H,CO-NH),δ 6.23~6.21(m,2H,Alkene),δ 5.78~5.76(q,1H,J=5 Hz,Alkene),δ 5.04~5.03(m,7H,Position 1),δ 2.95~2.80(s,4H,-CH2-)。其中,5.04~5.03是β-CD葡萄糖单元上C1-H的质子峰;6.23~5.76是丙烯酰基中-CH=CH的质子峰;2.95~2.80是乙二胺基中亚甲基-CH2-的质子峰且为单峰,由于受羰基的影响,亚甲基较乙二胺中该处的亚甲基向低场位移[17]。由此可见,目标产物β-CD-6-EA已成功合成。
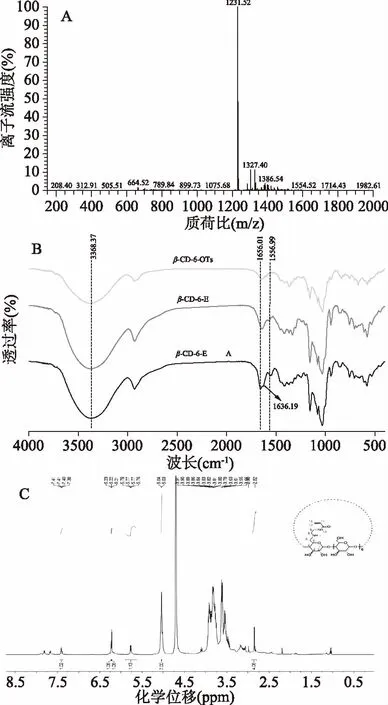
图3 β-CD-6-EA的质谱图(A)、傅立叶红外谱图(B)和核磁共振氢谱图(C)Fig.3 Mass,FTIR and 1H-NMR spectrum of β-CD-6-EA
2.3 β-CD-6-SA-AC的结构表征
β-CD-6-SA-AC的质谱结果如图4A所示:基峰对应的质荷比1411.33是由[β-CD-6-SA-AC(1410.48)+H(1)]+形成的;1357.33是由[β-CD-6-SA(1356.01)+H(1)]+形成的,终产物以单取代的β-CD-6-SA-AC为主,但是混有少量β-CD-6-SA,这可能是由于反应物SA-AC不稳定,部分遇水分解引起的。
由β-CD-6-SA-AC的红外图谱如图4B:3367.13和1031.09 cm-1为环糊精的特征吸收峰,1647.33 cm-1(仲酰胺Ⅰ谱带)是由酰胺的C=O伸缩振动产的,1555.73 cm-1(仲酰胺Ⅱ谱带)是由N-H弯曲振动和C-N伸缩振动组合吸收产生的[15];1599.24,1502.68和1458.55 cm-1为苯环中不饱和C-C键的特征吸收峰,1748.17 cm-1是SA-AC中C=O的伸缩震动吸收峰,1415.88 cm-1为C=C伸缩振动吸收峰。综上所述,β-CD-6-E和SA-AC成功发生酰化反应。
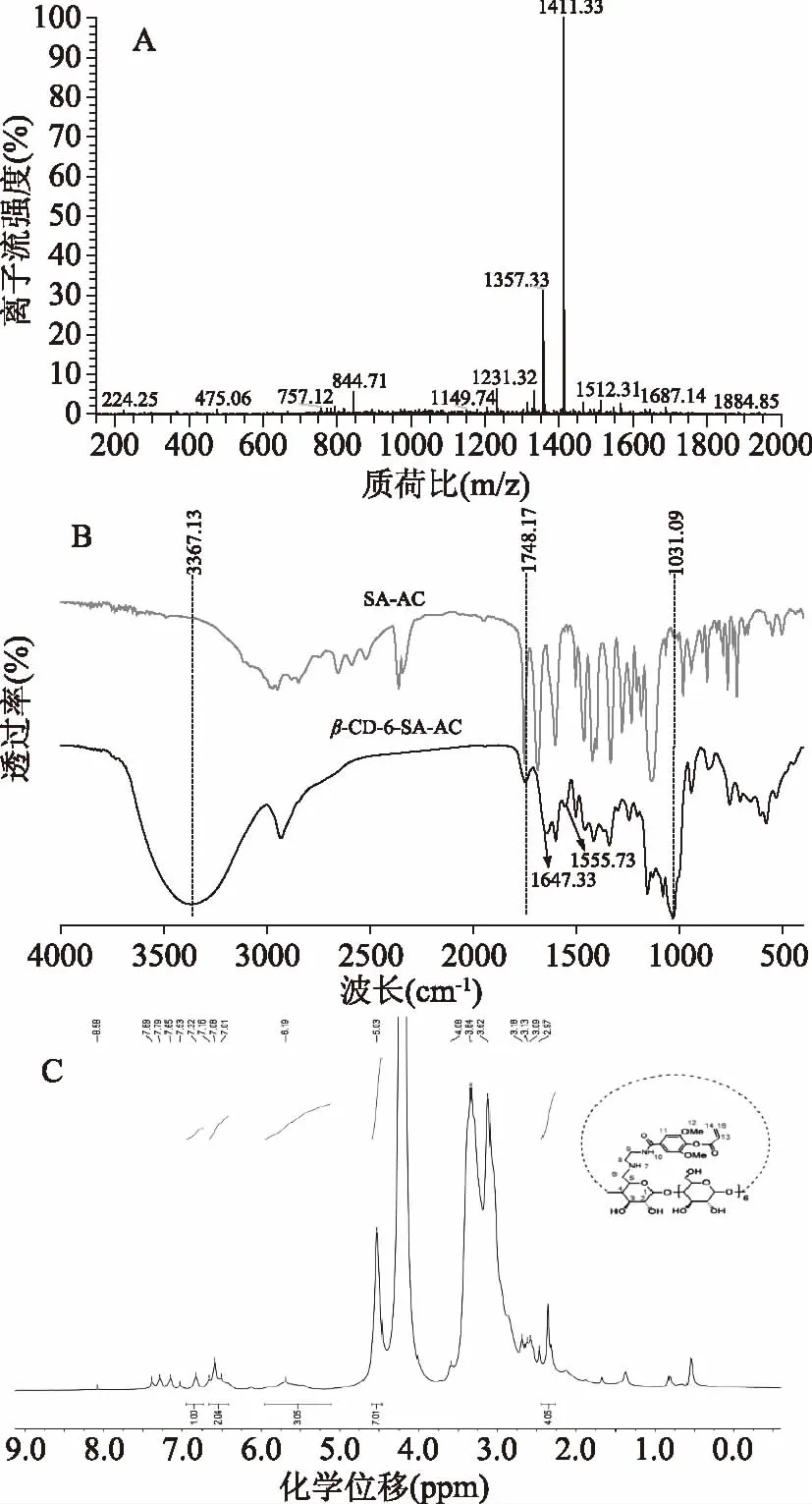
图4 β-CD-6-SA-AC的质谱图(A)、傅立叶红外谱图(B)和核磁共振氢谱图(C)Fig.4 Mass,FTIR and 1H-NMR spectrum of β-CD-6-SA-AC
采用1H-NMR对β-CD-6-SA-AC的结构进行表征,由4(C)可知:(500 Hz,D2O)δ 7.32(s,1H,CO-NH),δ 7.16~7.01(m,2H,Ar),δ 6.19~5.65(s,3H,Alkene),δ 5.03(s,7H,Position 1),δ 3.18~2.97(s,4H,-CH2-)。其中,5.03是β-CD葡萄糖单元上C1-H的质子峰且为单峰;7.16~7.01是苯环单元上C-H的质子峰,而苯环单元上-OCH3的质子峰应该表现在3.80左右,但由于与β-CD葡萄糖单元中其他位上-OH的质子峰重合,导致在δ 3.80出现一个很大的重叠峰;6.19~5.65是丙烯酰基中-CH=CH的质子峰;3.18~2.97是乙二胺基中亚甲基-CH2-的质子峰且为单峰。由此可见,目标产物β-CD-6-SA-AC已成功合成。
2.4 Allyl-β-CD的结构表征
Allyl-β-CD的质谱结果如图5A所示:较高峰对应的质荷比1197.52,1237.53,1277.49,1317.53分别是由[β-CD(1134.98)+Na(23)+40]+,[β-CD(1134.98)+Na(23)+2×40]+,[β-CD(1134.98)+Na(23)+3×40]+,[β-CD(1134.98)+Na(23)+4×40]+形成的。这说明终产物是具有不同取代度的Allyl-β-CD的混合物,但以双取代产物为主。
由Allyl-β-CD的红外图谱图5B:3392.16和1032.15 cm-1分别为环糊精O-H和C-O的振动吸收峰;1647.42 cm-1为C=C键的伸缩震动吸收峰,且在1597.30 cm-1附近出现C=C不对称伸缩震动吸收峰[19]。综上所述,烯丙基已经成功键合至β-CD分子中。
采用1H-NMR对Allyl-β-CD的结构进行表征,由图5C可知:(500 Hz,D2O)δ 5.38~5.22(m,3H,Alkene),δ 5.06(s,7H,Position 1)。其中,5.06是β-CD葡萄糖单元上C1-H的质子峰且为单峰;5.38~5.22是丙烯基中氢的质子峰;4.07~3.99是丙烯基上亚甲基-CH2-的质子峰,虽然与β-CD葡萄糖单元中其他位上-OH的质子峰混合在一起,但仍然可以分辨出[19]。由此可见,目标产物Allyl-β-CD已成功合成。
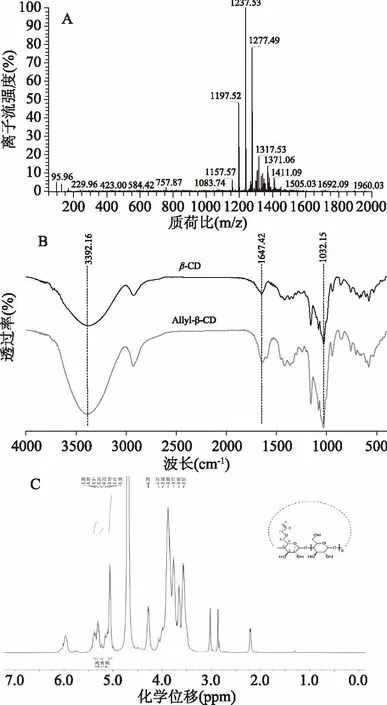
图5 Allyl-β-CD的质谱图(A)、傅立叶红外谱图(B)和核磁共振氢谱图(C)Fig.5 Mass,FTIR and 1H-NMR spectrum of Allyl-β-CD
3 结论
本文利用酰胺化反应合成单取代的β-CD-6-EA和β-CD-6-SA-AC,该方法具有反应彻底、产物易分离提纯、产率高的优点。具体制备条件:β-CD-6-E(1.0 eq)、AAc/SA-AC(1.2 eq)、HOBT(1.2 eq)、DIPEA(3.3 eq)、EDCI(1.5 eq),室温下反应12 h;利用亲核反应合成以双取代为主的Allyl-β-CD,该方法具有操作简单、耗时短的优点。具体制备条件:β-CD(1.0 eq)、NaH(20 eq)、3-溴丙烯(2.9 eq),0 ℃下搅拌1 h,室温下反应7 h。一方面,提高了含有双键的β-环糊精单体的制备效率;另一方面,丰富了含有双键的β-环糊精单体的种类,为今后制备各种功能的环糊精高聚物提供了应用基础和思路。
