偕二氟亚甲基苦马豆素类似物的设计与合成
2018-10-24张敏华李学辉蒋信义左玲玲
张敏华,李学辉,陈 晨,蒋信义,左玲玲,徐 军*
1.雅本化学股份有限公司,江苏 太仓 215433;2.沪东医院皮肤科,上海 浦东 201363
苦马豆素是澳大利亚学者Colegate[1]首先从灰苦马豆中分离得到的一种纯毒素。我国学者曹光荣等[2]亦从黄花棘豆中分离得到,并证实了其对α-甘露糖苷酶有很强的抑制作用。顾柏群等[3]进一步证实在茎直黄芪、变异黄芪中也含有苦马豆素。苦马豆素作为高效的α-葡萄糖苷酶抑制剂,不仅具有抗肿瘤作用[4],还具有抵抗病毒传染包括抑制HIV-1的效果[5]。因此,苦马豆素吸引了众多有机合成化学家和药物化学家的兴趣,已经有三十多条合成路线被报道[6-7],但对其构效关系的研究报道较少[8]。
然而,Tyler小组对与苦马豆素结构[图1(a)]相似的栗精胺的构效关系做了大量系统的研究工作。该小组在1995、1997年分别对栗精胺的C-8[9]、C-1和C-7位[10]羟基进行修饰后发现:栗精胺分子结构[图1(b)]中的C-1位羟基及其构型是影响其生物活性的关键位点,其手性是糖苷酶对氮糖识别的关键。该羟基的缺失会导致活性的丧失。而C-7位和C-8位的羟基对于生理活性的影响不是很大,对它们的改造虽会使分子的生物活性略有降低,但却能够提高分子对不同糖苷酶的抑制选择性。因此,将栗精胺C-7位和C-8位的羟基改造为其它基团或改变其手性,就可能寻找出选择性抑制不同类型糖苷酶的氮糖。

图1 (a)苦马豆素结构,(b)栗精胺结构Fig.1 Structures of(a)swainsonine,(b)castanospermine
对天然产物进行含氟改造是研究天然产物构效关系的重要手段之一。同时,将氟原子或含氟基团选择性地引入有机分子能显著改变原有分子的生理活性,可获得更多具有潜在生物活性的分子[11-12]。
鉴于此,对苦马豆素进行含氟及扩环改造,设计出了苦马豆素类似物1和2。其中,化合物1分子结构中保留了不饱和双键,在构象上可能会体现出和五元环类似的半椅式结构,这对研究苦马豆素的构效关系颇具意义(图2)。

图2 目标分子的设计Fig.2 Design of target molecules
关环复分解反应被认为是构建苦马豆素六元环最有效的方法[13-14]。通过分析目标分子的结构,六元环可以通过RCM反应来构建。关环前体3的五元环可以通过化合物4分子内亲核取代反应来形成,化合物4则可以通过关键中间体5′经过一系列转化得到,中间体5′的手性中心可以通过R-叔丁基亚磺酰胺的手性诱导产生[15](图3)。

图3 目标分子的逆合成分析Fig.3 Retrosynthestic analysis of target molecules
因此,对所设计的目标分子采用如下的合成路线(见图 4)。

图4 目标分子的合成路线Fig.4 Synthetic route of target molecules
1 实验部分
1.1 试剂与仪器
所用试剂均为国产市售分析纯。其中Grubbs'II催化剂(上海泰坦科技股份有限公司);乙酸乙酯三苯基溴磷盐为实验室自制。
Bruker AM-400核磁共振仪;Perkin-Elmer 241型自动旋光仪;温度计未校正。
1.2 化合物7的合成
将43.2 g R-叔丁基亚磺酰胺溶于120 mL二氯甲烷中,依次加入丙烯醛20.4 g、钛酸四异丙酯336.4 g,室温过夜。将上述反应液倒入200 mL冰水中,搅拌分散析出固体,硅藻土过滤,用二氯甲烷洗滤饼。分液,合并有机相。无水硫酸钠干燥,旋除溶剂,柱层析分离[m(PE)∶m(EA)=6∶1]得化合物748 g(产率 85%)[15]。
1.3 化合物5的合成
将38.4 g活化过的锌粉悬浮于干燥的THF(145 mL)中,加热至回流。缓慢滴加30 g化合物7和76.8 mL二氟溴乙酸乙酯的 THF(48 mL)溶液(反应比较剧烈,滴加要慢)。滴完后保温1 h,恢复至室温。加入饱和氯化铵溶液淬灭,分液,有机相饱和氯化钠洗涤,无水硫酸钠干燥,旋除溶剂,柱层析分离[m(PE)∶m(EA)=2∶1]得到化合物638.0 g(dr.=2.4∶1,产率 71%)[16]。
1H NMR(400 MHz,CDCl3)δ 5.97-5.89(m,0.35H),5.81-5.72(m,0.65H),4.42-4.26(m,3H),3.79(d,J=6.0 Hz,0.65H),3.48(d,J=9.6 Hz,0.35H),1.33(t,J=6.8 Hz,3H)1.20(s,6H),1.18(s,3H);13C NMR(100 MHz,CDCl3)δ 162.9(t,J=31.1 Hz),162.6(t,J=31.9 Hz),130.2(dd,J=3.1 Hz,1.5 Hz),129.3(t,J=3.8 Hz),123.0,122.0,113.9(t,J=255.8 Hz),113.8(t,J=255.8 Hz),63.4,63.1,61.5(dd,J=27.3 Hz,24.3 Hz),60.6(t,J=24.1 Hz),56.8,56.4,22.4,22.3,13.9,13.8;19F NMR (376 MHz,CDCl3)δ-110.7(dd,J=262.8 Hz,7.9F,0.3F),-113.5(dd,J=261.7 Hz,12.4Hz,0.7F),-114.9(dd,J=260.2 Hz,12.4 Hz,0.7F),-118.5(dd,J=261.3 Hz,16.2 Hz,0.3F)。
1.4 化合物8的合成
将24 g化合物7溶于360 mL二氯甲烷中,-78℃下滴加225 mL DIBAL-H(1.0 M in hex⁃ane)。滴毕,保温2 h。经点板跟踪,发现还有部分原料,补加DIBAL-H 40 mL,反应1 h。滴加饱和柠檬酸溶液淬灭反应。分液,有机相用饱和盐水洗涤,无水硫酸钠干燥。旋除溶剂后,将残留物溶于360 mL四氢呋喃中,加入23.4 mL三乙胺及36.8 g Ph3P+CH2CO2EtBr-,室温反应过夜。反应液经硅藻土过滤后旋干,粗产品经柱层析分离[m(PE)∶m(EA)=6∶1],得化合物812.6 g(产率:48%(两步))。
1H NMR(400 MHz,CDCl3)δ 6.79-6.69(m,1H),6.27-6.22(m,1H),5.65-5.40(m,1H),4.20-4.10(m,3H),3.66(d,J=4.0 Hz,1H),1.24(t,J=7.2 Hz,3H),1.16(s,9H);19F NMR(376 MHz,CDCl3)δ-105.8(dm,J=247.8 Hz,1F),-108.1(dt,J=151.2 Hz,11.3 Hz,0.8F),-108.5(dt,J=249.3 Hz,12.0 Hz,0.2F)
1.5 化合物9的合成
将2.5 g LiAlH4悬浮于干燥的四氢呋喃(25 mL)中,在0℃下滴加10 g化合物8的四氢呋喃(75 mL)溶液。滴毕,恢复到室温反应2 h。将体系降至0℃,缓慢加入饱和氯化铵溶液。粗产品经柱层析分离[m(PE)∶m(EA)=3∶1],得化合物96.27 g(产率:72%)。
1H NMR(400 MHz,CDCl3)δ 5.81-5.72(m,1H),5.47-5.40(m,2H),4.11-4.07(m,1H),3.73-3.72(m,1H),3.70-3.62(m,2H),2.14-1.94(m,2H),1.80-1.73(m,2H),1.22(s,9H);19F NMR(376 MHz,CDCl3)δ-101.5(dm,J=246.7 Hz,1F),-109.0(dm,J=245.2 Hz,1F)。
1.6 化合物10的合成
将6 g化合物9溶于60 mL二氯甲烷中,0℃下加入8.7 mL三乙胺、130 mg DMAP,滴加2.4 mL甲磺酰氯,恢复至室温反应3 h后加水淬灭反应。二氯甲烷萃取,有机相饱和盐水洗涤、无水硫酸钠干燥,过滤。将母液移至反应瓶中,降温至0℃,加入3.75 g叔丁醇钾,保温反应3 h。加水淬灭反应,有机相经1 mol/L HCl,饱和盐水洗涤,无水硫酸钠干燥。粗产品经柱层析分离[m(PE)∶m(EA)=8∶1],得化合物103.7 g(产率:66%)。
1H NMR(400 MHz,CDCl3)δ 5.91-5.73(m,1H),5.57-5.38(m,2H),3.96-3.88(m,1H),3.24-3.20(m,0.4H),3.14-3.11(m,1.2H),2.91-2.84(m,0.4H),2.05-1.62(m,4H),1.15(s,3H),1.13(s,6H);13C NMR(100 MHz,CD⁃Cl3)δ 129.9(dd,J=6.1 Hz,2.3 Hz),128.6(t,J=4.5 Hz),121.6,121.2,120.3(t,J=245.9 Hz),119.9(dd,J=248.8 Hz,242.9 Hz),66.6(dd,J=28.1 Hz,25.0 Hz),65.6(t,J=27.3 Hz),59.2,58.7,39.5,37.7,30.1(dd,J=46.3 Hz,23.6 Hz),29.9(dd,J=82.7 Hz,36.4 Hz),23.1(t,J=4.5 Hz),22.9,22.7,22.2(t,J=4.6 Hz);19F NMR(376 MHz,CDCl3)δ -110.1(m,0.75F),-100.9(dm,J=239.9 Hz,0.64F),-105.9(d,J=239.5 Hz,0.61F)。
1.7 化合物3的合成
将2 g化合物10溶于20 mL甲醇中,在0℃下加入28 mL饱和HCl/MeOH溶液,恢复至室温反应2 h,旋除溶剂。将所得油状物溶于150 mL DMF中,在0℃下往体系中加入6.7 g三乙胺、3.9 g氰基磷酸二乙酯和2.1 g 3-丁烯酸。恢复至室温反应过夜。加质量分数10%NaHCO3水溶液淬灭反应,二氯甲烷萃取3次,有机相饱和盐水洗涤,无水硫酸钠干燥。粗产品经柱层析分离[m(PE)∶m(EA)=8∶1],得化合物 31.12 g(产率:65%(两步)[17]。
1H NMR(400 MHz,CDCl3)δ 5.98-5.88(m,1H),5.84-5.73(m,1H),5.55(s,0.5H),5.39(dd,J=34.0 Hz,10.0 Hz,1H),5.27-5.10(m,3H),4.57(t,J=15.2 Hz,1H),3.67(d,J=13.2 Hz,0.4 H),3.19-3.06(m,2.5H),2.70-2.67(m,0.6H),2.06(s,1H),1.80-1.71(m,3H);13C NMR(100 MHz,CDCl3)δ 171.0,170.3,131.0,128.8,128.7,119.9,119.4,118.2,118.1,121.7(d,J=244.4 Hz),121.6(d,J=243.7 Hz),61.1(t,J=28.8 Hz),56.1(t,J=28.9 Hz),40.5,39.0,38.2,36.0,29.6,29.4,29.1,22.6,21.6(d,J=7.8 Hz),17.7,14.1;19F NMR(376 MHz,CDCl3)δ-100.3(dm,J=241.0 Hz,0.54F),-101.2(0.46F),-101.4(0.46F),-101.6(d,J=177.1 Hz,0.54F)。
1.8 化合物1的合成
将260 mg化合物3溶于60 mL甲苯中,加入130 mg Grubbs'II催化剂,加入回流反应过夜。冷至室温,旋干,直接柱层析分离[m(PE)∶m(EA)=1∶1],得化合物 1 170 mg(产率75%)[18]。
1.9 化合物2的合成
将50 mg化合物1溶于8 mL甲醇中,加入0.8 g m(Pd/C)(质量分数10%,包含质量分数50%的水)、甲酸2 mL,密闭反应48 h后过滤,旋干直接柱层析[m(MeOH)∶m(DCM)=1∶15],得化合物2为36 mg(产率70%)。
2 结果与讨论
根据合成分析,首先由R-叔丁基亚磺酰胺6与丙烯醛反应制备了R-叔丁基亚磺酰亚胺7。随后,R-叔丁基亚磺酰亚胺与现场生成的二氟溴乙酸乙酯锌试剂发生Reformatskii加成反应生成酯5。淬灭反应后,体系经氟谱检测,非对映异构体质量比例约为2.4∶1,两者不能通过柱层析分离。
Reformatskii加成反应主产物的构型可以通过如下六员过渡态来推导[19],化合物5'是加成反应的主产物,其构型也恰巧是我们所需要的(见图5)。
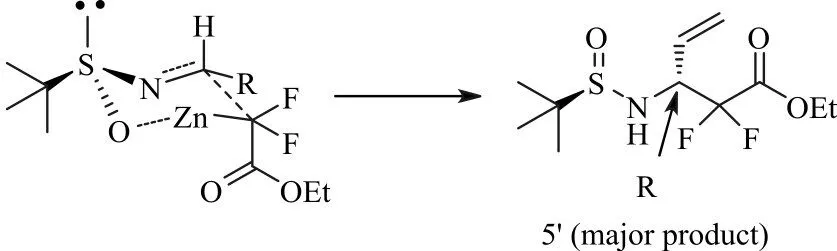
图5 化合物5'的构型Fig.5 Configuration of compound 5'
随后,对六元哌啶环进行构建。从化合物5'出发,DIBAL-H还原其结构中的a,b-不饱和酯基,所得的醛直接进行Wittig反应得到a,b-不饱和酯8。两个非对映体混合物不能通过柱层析加以分离。LiAlH4还原8中的a,b-不饱和酯基,得到不能直接地通过柱层析加以分离非对映体的伯醇9。将伯醇9的伯羟基上Ms,所得到的化合物直接在叔丁醇钾的作用下发生分子内亲核取代反应,得到化合物10,从而成功构建了哌啶环。并且,化合物10可以方便地与另一个异构体通过柱层析加以分离。
化合物10在盐酸甲醇溶液中脱除亚磺酰基后与3-烯丁酸反应,得到关环前体3。化合物3在Grubbs二代催化剂的催化下顺利关环生成了化合物1。氢化还原1的双键后,得到了化合物2。
3 结 语
通过Reformatskii加成反应和RCM等反应得到了苦马豆素的偕二氟亚甲基类似物,将通过后续的抗肿瘤活性测试来研究苦马豆素的构效关系,并期望得到更多生物活性物质。
