Peptide aptamer-mediated modulation of prion protein α-cleavage as treatment strategy for prion and other neurodegenerative diseases
2018-10-22AntoniaN.Klein,EricaCorda,SabineGilch
Despite intensive research, most neurodegenerative diseases cannot be cured and for some of them no treatment is available to increase survival or quality of life. Among the latter are prion diseases, fatal and transmissible neurodegenerative diseases of humans and other animals. Examples are Creutzfeldt-Jakob disease (CJD) in man, bovine spongiform encephalopathy (BSE, also known as mad cow disease) in cattle, scrapie in sheep and goats, and chronic wasting disease (CWD) in cervids. Most human prion diseases manifest sporadically, but also genetic and infectious origins are known. Prions, the causal agent of prion diseases, are composed solely of protein, namely a misfolded isoform of the cellular prion protein PrPC, termed PrPSc(Prusiner, 1998; Scheckel and Aguzzi, 2018). They are transmissible within and between species. Human-to-human transmission can occur through medical procedures (e.g.,neurosurgery) and results in iatrogenic CJD (iCJD). The largest number of iCJD cases worldwide has been reported upon treating growth hormone deficits with prion-contaminated cadaveric pituitary-derived human growth hormones.Transmission of BSE from cows to humans is to date the only example of zoonotic prion transmission resulting in variant CJD (Scheckel and Aguzzi, 2018). Whether CWD is transmissible to humans is unknown but represents a current threat due to expansion of geographic distribution within and beyond North America and in light of a novel study indicating oral transmission to non-human primates.In this context,finding a treatment against prion disease is of primary importance.
Prion protein isoforms:PrPCis expressed in many tissues with the highest level in the central nervous system, mainly in neurons. The glycosylphosphatidylinositol (GPI)-anchored PrPCis attached to the plasma membrane in association with detergent-resistant microdomains. It consists of aflexible N-terminal part and a globular C-terminal domain with three α-helices and two short β-strands. Additionally,the C-terminal part of PrPChas two glycosylation sites and a disulfide bond. The physiological role of PrPCis largely unknown. There is evidence for involvement in signal transduction, Cu2+binding, and neuroprotective activity(Scheckel and Aguzzi, 2018). More recently, neuroprotective and pathologic functions of PrPCin other neurodegenerative diseases, e.g., Alzheimer’s disease (AD) and Parkinson’s disease (PD) were reported (Brody and Strittmatter,2018).
In contrast, PrPScmanifests with high β-sheet content. It accumulates in the central nervous system and eventually leads to neuronal death. The conversion of PrPCto protease-resistant, aggregation-prone PrPScis the initial event in the pathogenesis of prion diseases. According to the seeded nucleation model, a direct interaction between PrPCand PrPScis necessary for autocatalytic conversion of PrPCto infectious PrPSc.
Previous therapeutic strategies for prion diseases included altering the subcellular trafficking of PrPC, activating the degradation of PrPSc, or inhibiting the interaction between PrPCand PrPSc(Gilch et al., 2007b; Corda et al.,2018). None of these approaches led to an approved therapeutic drug to treat prion diseases.
Anti-prion effect of peptide aptamers:More recently,we developed a new approach to inhibit the conversion of PrPCto PrPScby using peptide aptamers (PAs) binding to PrPC(Gilch et al., 2007a; Corda et al., 2018). PAs are peptides integrated into a scaffold protein, which ensures them a high conformational stability and therefore, an improved binding affinity compared to native peptides. We used the bacterial thioredoxin A as a scaffold protein to display different 16mer peptides. Three PAs interacting with PrPCwere selected by yeast-2-hybrid screening of a combinatorial library. Analysis of their binding sites to PrPCrevealed that one of them, PA8, has only one binding site to PrPC,recognizing amino acid residues 100–120 (PrP100–120).This is the most conserved domain of PrP and is crucial for the conversion of PrPCto PrPSc. Moreover, PrP100–120 harbors one high affinity binding site for toxic amyloid β-oligomers (Aβo) which are associated with pathogenesis of AD. In this context, the Aβo-PrPCinteraction results in inhibition of long-term potentiation and activation of neuronal cell death (Brody and Strittmatter, 2018). By modelling the PrP100–120-PA8 complex in silico, three amino acids within PA8 (W46, V47, T51) were identified as targets for site-directed mutagenesis to improve binding properties. Three out of eight PA8 derivatives (designated 46K,46Q, 47H) showed a maintained or improved inhibition of conversion of PrPCto infectious PrPSccompared to the lead PA8 upon treatment of prion-infected neuronal cells. This effect was dose-dependent and confirmed for different prion strains. Additionally, the inhibition of prion conversion could be maintained after treatment following removal of the PA and further passaging of neuronal cells. Furthermore, the here used PA inhibited de novo prion infection of neuronal cells. These results demonstrate the successful optimization of the PrP-PA interaction and subsequent improved inhibition of prion propagation. By analyzing their mode of action in more detail, we revealed that treatment of cultured cells with these PAs increases α-cleavage of PrPC(Corda et al., 2018).
PrPC α-cleavage: An alternative approach for the treatment of prion diseases is to enhance the physiological proteolytic cleavage of PrPC. However, proteolytic processing of the cellular prion protein is not totally understood.Until now, three main proteolytic cleavage events have been described: physiological α-cleavage at amino acids 110–111/112 (Figure 1), β-cleavage at amino acids 89/90 executed by calpains upon oxidative stress, and shedding at amino acids 228/229 mainly by the zinc metalloproteinase ADAM10 (Béland and Roucou, 2014). Here, we will focus only on α-cleavage.
Depending on the cell type and brain region, up to 50% of PrPCundergoes α-cleavage (Chen et al., 1995).α-cleavage occurs intracellularly in acidic endosomal compartments, the late secretory pathway or at the plasma membrane. It results in release of a soluble N-terminal fragment (~11 kDa, termed PrPN1) into the extracellular space and a membrane-bound C-terminal fragment (~17 kDa, termed PrPC1; Figure 1). However, the identity and proteolytic mechanism of the protease responsible for α-cleavage, termed α-PrPase, is still unclear. Some evidence indicates the involvement of ADAM10 for constitutive cleavage and ADAM17 activity upon simulation by agonists of the protein kinase C pathway. High levels of the cleavage product PrPC1 correlate with high levels of ADAM10 in the human brain. In contrast, ADAM10 knockout mice show normal α-cleavage in neurons. This indicates the involvement of more than one α-PrPase.PrPN1 has a neuroprotective role by reducing p53-dependent cell death in vitro and in vivo. PrPC1 has an increased stability and persistence at the cell surface. Notably, it cannot be converted to PrPScand moreover, acts as a dominant-negative inhibitor of PrPScformation. Therefore, enhancing α-cleavage represents a valuable treatment target for prion diseases and possibly other neurodegenerative diseases that benefit from high levels of neuroprotective PrPN1 and/or proteolysis of PrPCat the hydrophobic domain (Béland and Roucou, 2014). However, this approach is challenging due to the unknown identity of the α-PrPase.
Several studies indicate though that proteolytic processing of PrPCin the flexible N-terminal domain can be enhanced by structural stabilization. For example, structural stabilization of the N-terminal octapeptide repeat region increases β-cleavage (Lau et al., 2015). For α-cleavage, the hydrophobic domain (HD) (amino acids 111–129) is essential. Interestingly, the HD is also critical for physiological dimerization of PrP, which is linked to its stress protective activity (Rambold et al., 2008). Additionally, homodimerization facilitates PrPCtrafficking through the secretory pathway to the cell membrane. It was detected in human,bovine, mouse and hamster brain under experimental conditions and in N2a cells expressing hamster or endogenous PrPC. Notably, dimerization of PrP leads to increased α-cleavage, which indicates that protein binding to the HD may represent a possible approach to regulate α-cleavage,possibly by stabilizing the structure of this domain to favor α-PrPase cleavage.
Using PAs to modulate α-cleavage:The PAs we used in our study increased α-cleavage of PrPCand interfered with its internalization (Corda et al., 2018). We propose a mechanism similar to enhancement of α-cleavage upon PrP dimerization. Binding of PA to PrPCat amino acid 100–120 may lead to stabilization of theflexible N-terminus into a defined structure, which is more efficiently accessible for α-PrPase (Figure 1). The interference in internalization results in an increase of total PrPCand/or PrPC1 at the cell surface (Shyng et al., 1993). Given that α-cleavage can occur at the plasma membrane, prolonged presence of PrPCat the cell surface upon PA binding may in addition augment its exposure to α-PrPase. As a result, high levels of PrPC1 act as a negative inhibitor for prion conversion. Due to the increased α-cleavage, neuroprotective PrPN1 is generated and excreted into the extracellular space.
Taken together, physiological dimerization of PrPCleads to an increased α-cleavage and enhances neuroprotective activities. We propose that PA binding mimics the consequences of PrP dimerization. This enables us to increase α-cleavage and the production of PrPN1 and PrPC1 fragments. Using this approach, we circumvent the necessity of characterizing α-PrPase when attempting to enhance PrP α-cleavage as an approach for treatment of prion diseases.
Influence of α-cleavage in AD and synucleopathies:More recently, different studies revealed an involvement of PrPCin the pathogenesis of other neurodegenerative diseases. The amyloid cascade hypothesis has postulated that the deposition of the amyloid-β peptide is the causative agent for AD. Nowadays, the accumulation of the amyloid-β peptide in the nervous tissue is considered a key factor in the progression of this neurodegenerative disorder(Hardy and Higgins, 1992). The development of AD highly correlates with the level of Aβo, the most toxic species of self-aggregating Aβ. PrPChas two binding sites for Aβo: a low affinity site at amino acids 23–27 and a high affinity site at amino acids 95–110. The role of the PrP-Aβo-interaction is controversially discussed, but convincing evidence from different research groups indicates that PrPCacts as a receptor for Aβo and mediates the activation of toxic signaling pathways. This eventually leads to neuronal cell death, hyper-phosphorylation of tau as well as inhibition of long-term potentiation and impaired memory function in AD mouse models (Brody and Strittmatter, 2018). Our PAs bind to PrP100–120 suggesting that PA8 and its variants may competitively inhibit the PrP-Aβo interaction.
As mentioned, the Aβo binding site on PrPCis located in the PrPN1 fragment and the Aβo-induced toxic signaling requires GPI-anchored PrPC. Enhancing the α-cleavage at PrP residues 110–111/112 leads to an increased release of neuroprotective PrPN1 and consequently diminishes the Aβo ability to induce cell death.
Furthermore, the released neuroprotective PrPN1 acts as a decoy receptor for Aβo and competitively inhibits binding of Aβo to GPI-anchored full length PrPC. Notably, inhibition of Aβo-induced cell death after increased α-cleavage of PrPCin both cultured murine hippocampal neurons and in vivo has been reported (Brody and Strittmatter, 2018).
Another example for the influence of PrPCin a neurodegenerative disease are synucleopathies such as PD, associated with accumulation of misfolded α-synuclein in Lewy bodies (De Cecco and Legname, 2018). PrPCpromotes uptake of α-synuclein fibrils through a direct interaction with its N-terminal region. Moreover, the α-synuclein-PrPCinteraction at PrP amino acids 93–109 induces phosphorylation of Fyn kinase via metabotropic glutamate receptor 5, activates NMDA receptor and alters calcium homeostasis, which eventually leads to synaptic impairment. In neuronal cell lines incubated with α-synuclein fibrils, an increased α-cleavage of PrPCwas detected. Whether the increased α-cleavage of PrPCresults in a reduced uptake of α-synuclein is not reported, but this appears likely due to the N-terminal binding site of α-synucleinfibrils to PrP. In this scenario, again PA treatment to increase α-cleavage of PrP can be beneficial to prevent uptake, spread and toxic signaling of α-synucleinfibrils.
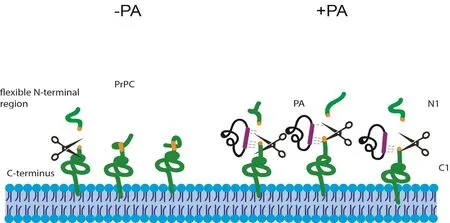
Figure 1 Alpha-cleavage of PrPC and the proposed mechanism of enhancement of PA binding.
Conclusion:In summary, a huge body of evidence demonstrates the central role of PrPCin the pathogenesis of prion and prion-like diseases, where it acts as a receptor for pathological protein aggregates and a mediator of toxic signaling. The PAs binding to PrPCwhich we have described here increase PrP α-cleavage. The resulting PrPN1 fragment is neuroprotective and can act as a decoy receptor for Aβo or a-synucleinfibrils, whereas the PrPC1 fragment lacks the interaction site and cannot transmit toxic signals. Moreover, it exhibits transdominant negative inhibition of prion replication. In light of these evidences,PAs targeting the PrP HD can be valuable tools and a novel approach towards treatment of multiple neurodegenerative disorders.
This work was funded by grants from the Alberta Prion Research Institute, the Alzheimer Society of Alberta and Northwest Territories and the Natural Sciences and Engineering Research Council (NSERC) of Canada. SG is supported by the Canada Research Chair program, ANK received a postdoctoral fellowship from the German Research Foundation (DFG).
Antonia N. Klein, Erica Corda, Sabine Gilch*
Department of Ecosystem and Public Health, Faculty of Veterinary Medicine, University of Calgary, Calgary,Alberta, Canada; Calgary Prion Research Unit, University of Calgary, Calgary, Alberta, Canada; Hotchkiss Brain Institute, University of Calgary, Calgary, Alberta, Canada
*Correspondence to:Sabine Gilch, PhD, sgilch@ucalgary.ca.
orcid:0000-0001-5923-3464 (Sabine Gilch)
Received:2018-05-29
Accepted:2018-07-18
doi:10.4103/1673-5374.241460
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Joaquin Martí-clúa, Universidad Autonoma de Barcelona, Spain.
Additionalfile:Open peer review report 1.
杂志排行

中国神经再生研究(英文版)的其它文章
- MicroRNAs of microglia: wrestling with central nervous system disease
- Huangqinflavonoid extraction for spinal cord injury in a rat model
- Apomorphine effects on the hippocampus
- Roles and functions of Atp6ap2 in the brain
- Magnesium sulfate and fetal neuroprotection:overview of clinical evidence
- Polyphenols-gut microbiota interplay and brain neuromodulation