MicroRNAs of microglia: wrestling with central nervous system disease
2018-10-22XiaoHuaWangTianLongWang
Xiao-Hua Wang , Tian-Long Wang ,
1 Department of Anesthesiology, Xuanwu Hospital, Capital Medical University, Beijing, China
2 Institute of Geriatrics, Beijing, China
3 National Clinical Research Center for Geriatric Disorders, Beijing, China
Abstract Microglia serve as brain-resident myeloid cells that affect cerebral development, ischemia, neurodegeneration, and neuro-viral infection. MicroRNAs play a key role in central nervous system disease through post-transcriptional regulation. Indeed, evidence shows that microRNAs are one of the most important regulators mediating microglial activation, polarization, and autophagy, and subsequently affecting neuroinfl ammation and the outcome of central nervous system disease. In this review, we provide insight into the function of microRNAs, which may be an attractive strategy and influential treatment for microglia-related central nervous system dysfunction. Moreover, we comprehensively describe how microgliafight against central nervous system disease via multiple functional microRNAs.
Key Words: microglia; neurodegeneration; central nervous system disease; microRNAs; activation; polarization;autophagy; neural regeneration
Introduction
Microglia are brain-resident myeloid cells with important functions in immune surveillance, mediating innate immune responses in the central nervous system (CNS). As a critical driver of neuroinflammatory responses, microglia secrete various substances that affect other glial cells and neurons, contributing to development of many neurological disorders such as cerebral ischemia, injury, neurodegenerative diseases, and neuroviral infections through microRNA(miRNA)-dependent regulation (Freilich et al., 2013). MiRNAs can control the magnitude of pathological conditions associated with neuroinflammation, as well as activating microglia by modulating the fate of different genes (Guedes et al., 2013). At least eight miRNAs are uniquely, highly expressed in microglia versus other immune and myeloid cells(Butovsky et al., 2014). MiRNAs provide a novel layer of microglial gene regulation, leading to new treatment opportunities for microglia-associated CNS dysfunction (Table 1).
MicroRNAs Modulate Microglial Polarization
Microglia in normal CNS exhibit a resting (ramified) M0 phenotype expressing CD11b. Interferon (IFN)-γ/lipopolysaccharide (LPS) stimulates microglia to a classical‘M1’-skewing phenotype, which enhances expression of pro-inflammatory cytokines. MiR-124, miR-155, and miR-689 are most strongly associated with a M1-like phenotype,and these miRNAs mediate pro-inflammatory pathways. Interleukin (IL)-4/IL-13 changes microglia to a ‘M2a’-skewing phenotype for anti-inflammatory and tissue repair functions(Orihuela et al., 2016). MiR-711 and miR-145 strongly mediate a M2-like activation phenotype and anti-inflammatory pathway function (Veremeyko et al., 2012). MiR-124 is expressed in microglia but not peripheral monocytes or macrophages. Brain-specific miR-124 directly and negatively modulates CCAAT/enhancer-binding protein-α (C/EBP-α)transcription factor, and subsequently, the downstream C/EBP-α target, PU.1, leading to transformation of microglia from an activated to quiescent phenotype. In this state, a major histocompatibility complex (MHC) class II phenotype is expressed, resembling resting microglia (Ponomarev et al.,2011; Zhang et al., 2017). Intrathecal miR-124 injection can treat and prevent persistent microglial activation, restoring persistent hyperalgesia, and decreasing the ratio of M2/M1 microglial type markers in the spinal cord of G protein-coupled receptor kinase 2 (GRK2)-deficient mice (Willemen et al., 2012). Anti-miR-155 treatment can prolong microglial survival. In superoxide dismutase 1 (SOD1) mice, miR-155 expression increases loss of the microglial homeostatic molecular signature, and consequently, microglial biological functions (including phagocytosis) are suppressed(Moore et al., 2013). Upregulated miR-9 specifically induces transgene-expressing cells with typical ramified microglial morphological features, with the majority of transgene-expressing cells labeled by the microglial marker, ionizedcalcium binding adaptor molecule 1 (Iba1). Monocyte chemotactic protein-induced protein 1 (MCPIP1) is crucial for controlling inflammation in the nuclear factor (NF)-κB pathway. MiR-9 plays a unique role in mediating microglial inflammatory responses by downregulating MCPIP1 expression (Akerblom et al., 2013; Yao et al., 2014). After LPS treatment, IL-6 expression was significantly increased in microglia transfected with miR-101a. MiR-101a enriched in the brain also contributes to differentiation of bone marrow cells into microglia-like cells (Saika et al., 2017).
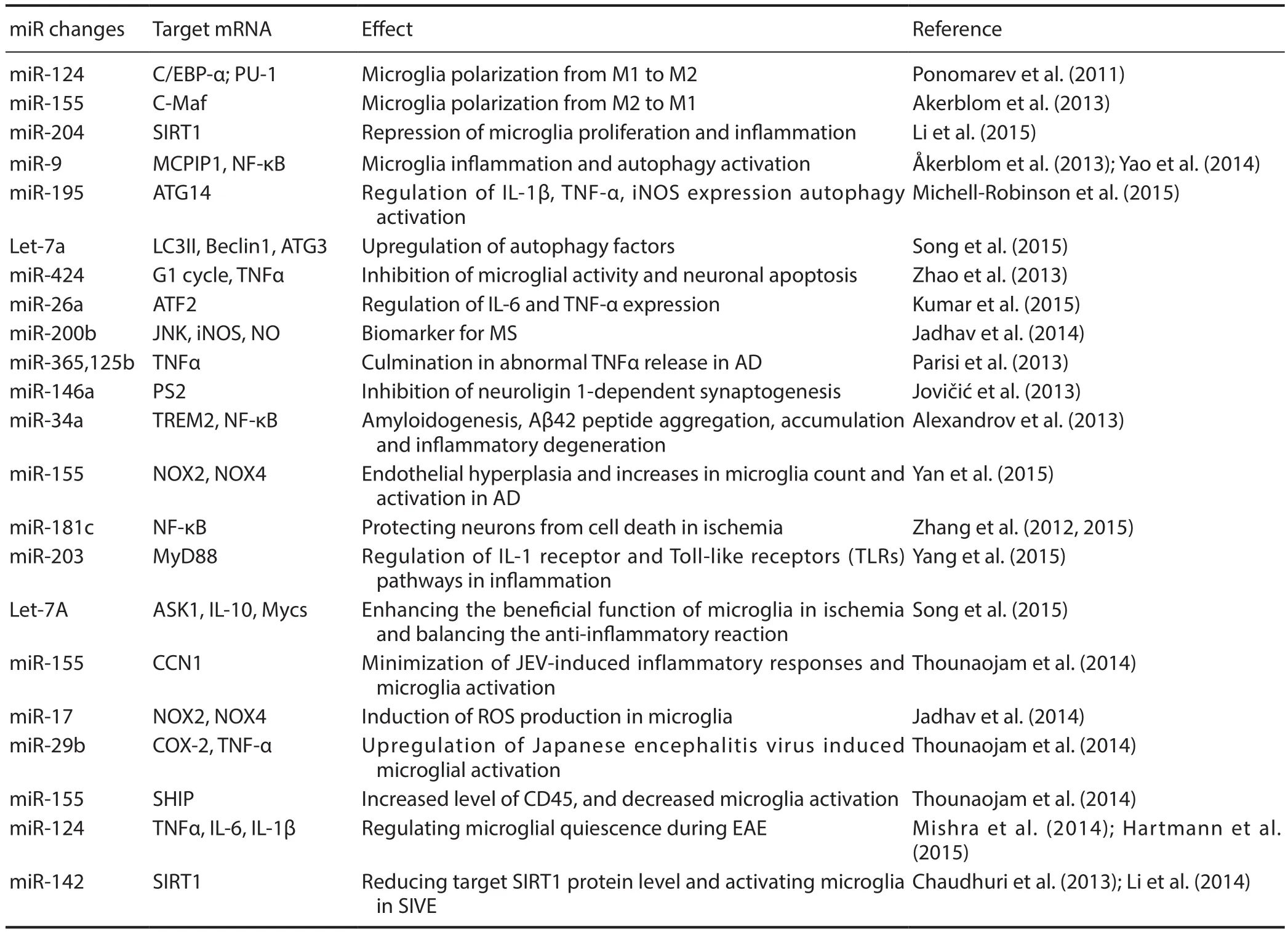
Table 1 Changes of microRNA (miR) in microglial regulation
MicroRNAs Involved in Autophagy of Microglia and CNS Development
Microglia serve as critical nervous system-specific immune cells that influence brain development and maintain brain function in both healthy and pathological contexts. Microglia-enriched miRNAs inhibit aberrant glial expression of neuronal phenotypes and proteins. In microglia, Let-7a is involved in increased expression of brain derived neurotrophic factor (BDNF), IL-10, IL-6, and IL-4. Let-7a also reduces nitrite production and inhibits expression of inducible nitric oxide synthase (iNOS), which ultimately improves neuronal development (Cho et al., 2015). MiR-204 can repress inflammatory processes and microglial proliferation, and promote microglial apoptosis via increased sirtuin 1 (SIRT1) expression (Li et al., 2015). Upregulated miR-424 decreases microglial activity, further causing G1 phase cell-cycle arrest and neuronal apoptosis. MiR-424 is also involved in reducing tumor necrosis factor (TNF)-α production and suppressing ionized calcium binding adaptor molecule-1 (Iba1) immunoreactivity and protein levels following induction of neuron development (Zhao et al.,2013a). During development, the miR-155/CCN1 regulatory axis balances proinflammatory and proangiogenic activities of microglia. Function of miR-155 on the microglial target,CCN1, mediates anastomosis and sprout fusion, thereby modulating inflammation-induced vascular homeostasis and repair (Yan et al., 2015). Compared with other myeloid lin-eage cells, microglia represent a distinct cell population that enters the CNS under pathological conditions to perform an autophagic function (Michell-Robinson et al., 2015).Microglial autophagy is the pivotal process for cytokine production and cellular survival. At least four miRNAs are involved in the microglial autophagy system. Recent studies in cultured microglia have shown that miR-146a inhibits neuroligin 1-dependent synaptogenesis (Jovicic et al., 2013).Similarly, downregulated miR-195 can increase activation of microglial autophagy. Accordingly, inhibition of miR-195 expression suppresses neuroinflammation and neuropathic pain (via IL-1β, iNOS, and TNF-α) through 3′UTR targeting of autophagy related 14 (ATG14) (Shi et al., 2013). As a key modulator, particularly in the autophagy system, miR-Let7A enhances autophagy efficiency in various inflammatory responses and activates microglia. In LPS-stimulated microglia, Let-7a upregulates expression of autophagy-related factors such as Beclin1, ATG3, and microtubule-associated protein 1A/1B-light chain 3 (LC3) (Song et al., 2015). After LPS stimulation in microglia, miR-26a expression is rapidly reduced. Over-expression of miR-26a decreases TNF-α and IL-6 via a mechanism independent of activating transcription factor 2 (ATF2) (Kumar et al., 2015). Intracerebral hemorrhage mediates microglial autophagic activation. The miR-144 target on target of rapamycin (TOR) regulates microglial inflammation and autophagic reaction (Wang et al.,2017).
MicroRNAs of Microglia Contribute to Development of Neurodegenerative Diseases
Microglia contribute to modifying neuroinflammatory process in Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS) (Parisi et al., 2013; Shadfar et al., 2015; Li et al., 2016; Fan and Pang, 2017).
After treatment with miR-200b inhibitor, neuronal apoptosis was significantly increased and neurodegeneration aggravated (Jadhav et al., 2014a). MiRNA-200b induces migratory potential of activated microglia by decreasing c-Jun N-terminal kinase (JNK) activity and iNOS and nitric oxide(NO) production. Chronic activation of microglia induces neuronal damage due to excessive release of neurotoxic molecules and proinflammatory cytokines in AD. MiR-155 is one of the most well studied immune-related miRNAs,and its overexpression increases microglial activation and contributes to AD-associated neuroinflammation (Guedes et al., 2014). In AD brain, miRNA-34a transfection-mediated downregulation of triggering receptor expressed on myeloid cells 2 (TREM2) target impaired phagocytic responses that ultimately contribute to amyloidogenesis, amyloid-β (Aβ)-42 peptide aggregation and accumulation, and inflammatory degeneration (Alexandrov et al., 2013). Presenilin 2 (PS2) is a membrane associated protease that regulates proinflammatory microglial behavior. Hence, PS2 deficiency or dysfunction may result in uncontrolled pro-inflammatory activation contributing to AD neurodegeneration. As a negative regulator of pro-inflammatory responses in knockdown microglia, miR-146 constitutively downregulates PS2. MiR-146 has been implicated in the pathogenesis of AD by limiting the pro-inflammatory response (Jayadev et al., 2013).In the SOD1-G93A mouse model, miR-125b and miR-365 culminate in abnormal TNF-α release from primary microglia, thus contributing to AD as a vicious gateway (Parisi et al., 2013). ALS is a progressive disease targeting motor neurons and neighboring glia that is associated with microgliosis and a decrease in resident microglia, which directly contributes to neurodegeneration and final death within 5 years (Butovsky et al., 2012). Microglia are involved in ALS toxicity and neuronal protection. Some miRNAs contribute to the pathogenesis of ALS. Up-regulated miR-125b leads to an uncontrolled toxic M1 microglial reaction (Parisi et al.,2016). The miRNA-34a target of TREM2 expression is mediated by the NF-κB pathway and involved in phagocytic and innate immune responses that contribute to inflammatory neurodegeneration in ALS (Zhao et al., 2013b). Following transfection of NSC34 cells with SOD and co-culture with(N9) microglial cells, miR-124 increased in both NSC34 cells and the transfer exosomes. Consequently, treating microglia with these exosomes can induce a switch from a mixed M1 and M2 subpopulation to M1 polarization, shown by co-expression with IL-1β, iNOS, MHC-II, and TNF-α. Indeed,miR-124 may become a potential and powerful therapeutic strategy for providing benefit to ALS patients by halting microglial activation (Mishra et al., 2014).
MicroRNAs Play Key Roles in Microglial Activation in Response to Brain Ischemia and Injury
CNS ischemia and injury often induce microglial activation and are associated with release of inflammatory cytokines(Wang et al., 2016). Activated microglia have multiple effects on glia and neurons by secreting various substances (Cho et al., 2015). Understanding how miRNAs alter the microglial transcriptome may provide valuable insight into how activated microglia mediate neurological damage following ischemia and injury. As a model of ischemic insult, oxygen/glucose deprivation (OGD) induces microglial activation(as shown by overproduction of TNF-α, IL-1β, and NO),and modulates expression of inflammatory-related and brain-enriched miRNAs (Kong et al., 2014). In primary microglia, hypoxia downregulates miR-181c. OGD can activate microglia and subsequently cause neuronal death. However, ectopic upregulated miR-181c protects neurons against damage by inhibiting TNF-α mRNA at the 3′-untranslated region (3′UTR) (Zhang et al., 2012a). MiR-181c inhibits toll-like receptor (TLR)-4 expression and represses NF-κB activation and downstream pro-inflammatory mediators (Zhang et al., 2015). Thrombin can induce microglial activation,leading to downregulation of miR-181c. Upregulated miR-181c expression has an opposing effect on NF-κB activity and the pro-inflammatory response, subsequently inhibiting microglial activation through its target, mixed-lineage leukemia-1 (MLL1) (Yin et al., 2017b). In the developing brain and during neural regeneration, neuroinflammation mediated by activated microglia plays a critical role in the pathogenesis of hypoxic injury (Zhou et al., 2015). Transfection with miR-203 results in microglial activation by targeting MyD88, which is an important adapter protein regulating IL-1 receptor and TLR pathways during inflammation and ischemia (Yang et al., 2015). Let-7c-5p overexpression may suppress microglial activation and protect the brain from ischemic damage though the miRNA-modulated caspase-3 pathway (Ni et al., 2015). MiR-Let7a can enhance the beneficial function of microglia in ischemia, balance the anti-inflammatory cytokines, IL-10, c-Myc, and N-Myc,and activate apoptosis signal-regulating kinase 1 (ASK1)(Song and Lee, 2015). Thymosin β4 (Tβ4) works as a major G-actin-sequestering molecule. MiR-146a expression is increased after Tβ4 treatment, and suppression of microglial activation proceeds the anti-inflammatory effect following hypoxic brain injury (Zhou et al., 2015). Following hypoxic microglial activation, Fas ligand (FasL) overproduction mediates neuronal apoptosis, whereas miR-21 ectopic expression targets FasL and partially protects neurons from death caused by hypoxia-activated microglia (Zhang et al., 2012b).Constitutive miR-155-deficiency in mice contributes to mild vascular defects, while decreased miR-155 expression causes endothelial hyperplasia and increases microglial count and activation by inhibiting expression of CCN1 target. Following ischemic insult, knockout miR-155 inhibits microglial activation, normalizes retinal vasculature, and inhibits abnormal vascular growth in mice (Yan et al., 2015).
Microglia Defend against Neuro-Viral Infections by MicroRNA
Microglia are the primary defenders against neuro-viruses and pivotal to CNS remodeling. Various miRNAs regulate microglial activation during neuro-viral infections (Martinez and Peplow, 2017). Activated microglia mediate neuroinflammatory events in human immunodeficiency virus(HIV)-associated neurological disorders. MiR-17 induces reactive oxygen species (ROS) production in microglia by regulating NADPH oxidase (NOX)-2 and NOX4 targets during HIV neurological dysfunction. Increased ROS production subsequently induces cytokine production, and in turn, leads to microglial activation (Yan et al., 2015). Microglial activation is a critical event in Japanese encephalitis virus (JEV)-induced neuroinflammation. MiR-29b mimic diminished expression of pro-inflammatory cytokines and decreased iNOS and cyclooxygenase-2 levels in microglia.Knockdown of miR-29b upregulates JEV-induced microglial activation by overexpression of TNF-α-induced protein 3, and subsequently, decreased nuclear translocation of phosphorylated NF-κB (Jadhav et al., 2014b). MiR-155 expression was upregulated in primary microglia during JEV infection. Knockdown of miR-155 minimized JEV-induced inflammatory responses via negative regulation of SH-2 containing inositol 5′ polyphosphatase 1 (SHIP1) expression (Thounaojam et al., 2014a). Upon JEV infection, overexpression of miR-155 causes decreased expression of phosphorylated signal transducers and activators of transcription(p-STAT1). Moreover, miR-155 mimic can increase CD45 levels and decrease microglial activation (Thounaojam et al.,2014b). MiR-155 deficiency protects microglial cells (BV2 cells) from inflammation using LPS induced by targeting receptor for activated C kinase 1 (RACK1) (Yin et al., 2017a).Upregulated miR-142-3p and -5p expression within microglia in SIV encephalitis (SIVE) animals, reduces target SIRT1 protein levels (Chaudhuri et al., 2013; Li et al., 2014;Pareek et al., 2014). MiR-124 is a key regulator of microglial quiescence during experimental autoimmune encephalomyelitis (EAE). In activated microglia, miR-124 expression is decreased. Upregulated miR-124 alone significantly decreases IL-6, IL-1β, and TNF-α expression. In EAE, peripheral administration of miR-124 reduced activation of myelin-specific T cells, causing systemic deactivation of macrophages and markedly inhibiting disease development.MiR-124 itself is very important for maintaining microglial quiescence, decreasing the activity of several signaling pathways, and ultimately decreasing neuronal death and neurotoxicity (Mishra et al., 2014; Hartmann et al., 2015).
Summary
Dysregulated microglial behavior can result in inflammatory activation, microglial reaction, and subsequent neurological disorders (Figure 1). Further, the evidence supports the crucial role of miRNAs in regulating gene expression of microglia and microglial function. Thus, miRNA-based therapies are an attractive strategy for modulating microglial activity,and may affect the outcome of multiple CNS diseases in the future.
Author contributions:Study design and supervision: TLW; paper writing: XHW. Both authors approved thefinal version of this paper.
Conflicts of interest:There are no conflicts of interest associated with this manuscript.
Financial support:This work was supported by the National Natural Science Foundation of China, No. 81401084 (to XHW); Beijing Municipal Administration of Hospital Ascent Plan, No. DFL20150802 (to TLW); Beijing 215 High Level Healthcare Talent Plan Academic Leader, No. 008-0027 (to TLW); Beijing Municipal Commission of Health and Family Planning, No. PXM2017_026283_000002 (to TLW); Bei-jing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding, No. ZYLX201706 (to TLW).
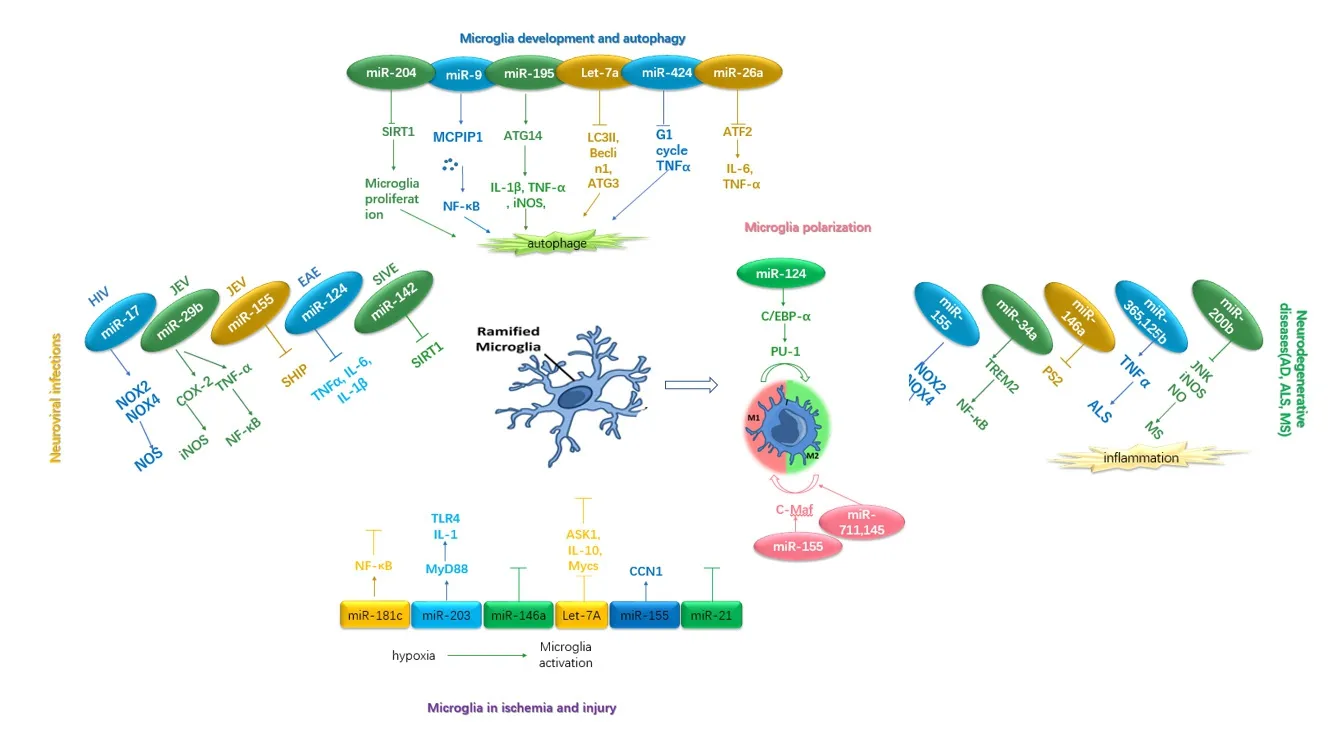
Figure 1 The relationship between microRNAs and microglia.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Ozgur Boyraz, Gulhane Military Medical Academy, Turkey; Shruti V. Kabadi, University of Maryland, USA.
Additionalfile:Open peer review reports 1 and 2.
杂志排行

中国神经再生研究(英文版)的其它文章
- Inhibiting the kynurenine pathway in spinal cord injury: multiple therapeutic potentials?
- Huangqinflavonoid extraction for spinal cord injury in a rat model
- Apomorphine effects on the hippocampus
- Roles and functions of Atp6ap2 in the brain
- Magnesium sulfate and fetal neuroprotection:overview of clinical evidence
- Polyphenols-gut microbiota interplay and brain neuromodulation