Inhibiting the kynurenine pathway in spinal cord injury: multiple therapeutic potentials?
2018-10-22KellyJacobsDavidLovejoy
Kelly R. Jacobs, David B. Lovejoy
Neuroinflammation Group, Centre for Motor Neuron Disease Research, Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, Macquarie University, Sydney, Australia
Abstract Chronic induction of the kynurenine pathway (KP) contributes to neuroinflammation by producing the excitotoxin quinolinic acid (QUIN). This has led to significant interest in the development of inhibitors of this pathway, particularly in the context of neurodegenerative disease. However, acute spinal cord injury(SCI) also results in deleterious increases in QUIN, as secondary inflammatory processes mediated largely by infiltrating macrophages, become predominant. QUIN mediates significant neurotoxicity primarily by excitotoxic stimulation of the N-methyl-D-aspartate receptor, but other mechanisms of QUIN toxicity are known. More recent focus has assessed the contribution that neuroinflammation and modulations in the KP make in mood and psychiatric disorders with recent studies linking inflammation and modulations in the KP, to impaired cognitive performance and depressed mood in SCI patients. We hypothesize that thesefindings suggest that in SCI, inhibition of QUIN production and other metabolites, may have multiple therapeutic modalities and further studies investigating this are warranted. However, for central nervous system-based conditions, achieving good blood-brain-barrier permeability continues to be a limitation of current KP inhibitors.
Key Words: spinal cord injury; neuroinflammation; kynurenine pathway; activated microglia; infiltrating macrophages; quinolinic acid; neuropsychiatry; depression
Neuroinflammation, Microglial Phenotype and the Kynurenine Pathway (KP)
The inflammatory response initially involves induction of various cytokines and chemokines to coordinate insult repair. However, in the context of the central nervous system(CNS), sustained neuroinflammation, results in a set of well-described neuroinflammatory events that exacerbate injury. Central to these processes is the switch from the neuroprotective M2 microglial/infiltrating macrophage phenotype to the neurotoxic M1 phenotype, mediated largely by interferon-γ (INF-γ) or lipopolysaccharide (LPS). It is well reported that activated M1 microglia and infiltrating macrophages are the prime generators of the increased quinolinic acid (QUIN) levels observed in all the major neurodegenerative conditions. In fact, macrophages stimulated by INF-γ produced nearly 40-fold more QUIN relative to microglia (Guillemin et al., 2004). QUIN potently agonizes the N-methyl-D-aspartate (NMDA) receptor, resulting in excitotoxic stimulation that initiates a cascade of events involving neuronal Ca2+influx, neuronal nitric oxide synthase(nNOS) activation that increases nitric oxide (NO) production and redox stress, which in-turn results in mitochondrial membrane destabilisation, activation of poly ADP-ribose polymerase (PARP), NAD+depletion and eventually neuronal cell death (Jacobs et al., 2017; Figure 1). Accordingly, excessive QUIN production by the over-activated KP is viewed as a significant neuroinflammatory contributor and inhibition of QUIN production has received much attention as a potential therapeutic strategy for neurodegenerative disease.Another potentially neurotoxic metabolite resulting from KP activation is 3-hydroxykynurenine (3-HK), produced by the enzyme kynurenine 3-monooxygenase (KMO) following hydroxylation of its substrate, kynurenine (KYN). Originally viewed as a neurotoxic metabolite that produces substantial redox activity, 3-HK is now thought to exert more pleiotropic effects on redox status (Jacobs et al., 2017). Similarly,like 3-HK, 3-hydroxyanthranilic acid (3-HAA) was shown to induce neuronal death by redox stress mechanisms, although it was also shown to exert anti-inflammatory effects by supressing cytokine and chemokine expression in activated glial cells (Krause et al., 2011). Other studies showed that while 3-HAA inhibited the Fenton reaction and scavenged reactive oxygen species (ROS) in a deoxyribose degradation assay, pro-oxidant effects were observed in an iron(II) autoxidation assay (Chobot et al., 2015). Interestingly,decreased 3-HAA expression is reported in Huntington’s disease, stroke and major depression and as 3-HAA is readily oxidized by radical scavenging, lower 3-HAA concentrations are considered a sensitive biomarker of ROS-mediated disease processes (Darlington et al., 2010).
Spinal Cord Injury (SCI), Macrophages and the KP
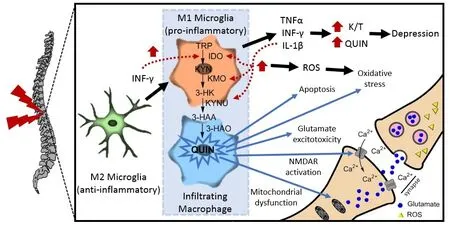
Figure 1 Kynurenine pathway (KP) involvement in spinal cord injury (SCI).
SCIs fail to heal like skin or muscle injuries. One perspective suggests that this reflects desynchronization of the conventional sequential pattern of macrophage phenotypes, in which the neuroprotective M2 phenotype that promotes proliferative and remodelling phases of repair,becomes dominated by the pro-inflammatory M1 phenotype such that remodelling is not properly initiated. These authors draw parallels to the chronic inflammation present in non-healing wounds (Gensel and Zhang, 2015). Indeed,shifting the cytokine profile towards that of the neuroprotective M2 phenotype by temporal blockage of interleukin(IL)-6 improved locomotor Basso Mouse Scale (BMS) score following SCI, reduced levels of INF-γ and tumor necrosis factor-α (TNF-α) and increased anti-inflammatory IL-4 and IL-13 at the site of the spinal lesion (Guerrero et al., 2012).Key proinflammatory drivers resulting from prolonged engagement of the M1 phenotype include TNF-α, IL-1β,IFN-γ, NO and superoxide anion (O2–), which are either known to activate the KMO branch of the KP (TNF-α, IL-1β and IFN-γ) or are products of KP activation (NO and O2–;Figure 1). Thus, KP induction, particularly when chronic,has the potential to engage a vicious-cycle that further fuels KP dysregulation and consequent damage. Considering the numerous observations of infiltrating macrophages in SCIs,the link to the KP becomes further compelling when it is appreciated that activated macrophages produce 204-fold more QUIN than non-activated macrophages and at levels identified as neurotoxic (Guillemin et al., 2004).
Hemeoxygenase-1 in SCI, Relationship to Nrf2 Signaling and the KP
Induction of the enzyme heme oxygensase-1 (HO-1) is frequently reported in animal models of SCI. For instance, in rat SCI, HO-1 was overexpressed by activated neutrophils and use of a HO-1 inhibitor delayed motor function recovery after SCI, suggesting that HO-1 coordinates significant anti-inflammatory and protective effects in damaged tissue(Liu et al., 2002). The nuclear factor erythroid 2-related factor 2/anti-oxidant response (Nrf-2/ARE) pathway is described as the master regulator of the cellular anti-oxidant defence network and is a known inducer of HO-1. Essentially, when exposed to ROS, Nrf-2 translocates to the nucleus,where it activates HO-1 transcription. Interestingly, the KP metabolites QUIN and 3-HK can both promote Nrf-2 nuclear translocation (Colin-Gonzalez et al., 2014a, b). This could imply an activity in opposition to their described deleterious roles in neurodegenerative disease. However, QUIN treated rat striatal slices showed increased Nrf-2 nuclear translocation after 1 hour and reduced lipid peroxidation after 3 hours (Colin-Gonzalez et al., 2014a). This temporal lag suggests that QUIN-induced nuclear translocation of Nrf-2 was not a direct effect, but rather a compensatory response to QUIN induced ROS production which forms part of the general anti-oxidant defence system. Although data is lacking, on balance, it seems likely that 3-HK also induces Nrf-2 translocation as a compensatory mechanism.However, unlike QUIN, 3-HAA seems to mediate an overall neuroprotective effect. In primary human fetal CNS cultures treated with IL-1 (with or without IFN-γ) or with Toll-like receptor ligands (as a model of the proinflammatory CNS environment), 3-HAA treatment suppressed glial cytokine and chemokine expression and reduced cytokine-induced neuronal death (Krause et al., 2011). 3-HAA also markedly increased the expression HO-1 by astrocytes and macrophages (Krause et al., 2011). After further in-vivo assessment, the authors concluded that 3-HAA showed promising activity as a potential neuroprotective compound.
Do Neuropsychiatric Symptoms Further Connect Macrophages and SCI to the KP?
There is also growing interest in the role that KP activation plays in major depression, suicidal ideation, autism spectrum disorder and schizophrenia (Steiner et al., 2011; Lim et al., 2016; Bryleva and Brundin, 2017; Plitman et al., 2017).Influential studies include those by Steiner et al. (2011), who showed that depressed patients had significantly increased immunoreactive QUIN-positive microglia in the subgenual anterior cingulate and cortex anterior midcingulate cortex compared to controls. In suicide attempters QUIN was significantly elevated approximately 2-fold in the cerebro-spinalfluid (CSF) relative to controls and was associated with increased CSF IL-6. QUIN CSF levels also correlated to scores on Suicide Intent Scale and significantly decreased 6 months post suicide attempt (Erhardt et al., 2013). Studies also link inflammation and KP modulation to mood disturbances in SCI patients. In a randomized, controlled clinical trial, 20 patients with varying severity of SCI and 20 control subjects received an anti-inflammatory diet for 3 months(Allison and Ditor, 2015). Significant group × time interactions were found for standardized score of depression, the ratio between tryptophan (TRP) and large neutral amino acids (LNAA; that reflects brain TRP concentration) and the composite score of pro-inflammatory cytokines. Changes in IL-1β significantly correlated to standardized depression score and kynurenine/tryptophan (K/T) ratio (Pearson’s r correlations). Changes in K/T ratio were also significantly correlated with depression scores, with further statistical analysis showing that this correlation was mediated by changes in IL-1β (Allison and Ditor, 2015). Although a subsequent follow-up study showed that there were no significant links between consumption of an anti-inflammatory diet in SCI to improvements in cognitive function (Allison et al., 2017), presumably because participants had underlying hippocampal damage not tractable by reduced inflammation.
Targeting the KP in SCI
An early report showed that after a brief compression injury to the lower thoracic spinal cord (T13) of adult guinea pigs,QUIN levels were elevated greater than 10-fold at the injury site after 5 days, while the activity of the first enzyme in the KP indoleamine 2,3-dioxygenase (IDO-1) was increased approximately 2-fold. These changes were not found in adjacent undamaged regions (Blight et al., 1993). Other studies have demonstrated that injections of 4-chloro-3-hydroxyanthranilate (4-Cl-3-HAA) a 3-hydroxyanthranilic acid oxygenase(3-HAO) inhibitor, blocking the metabolism of 3-HAA into QUIN, every 12 hours for 12 days post SCI significantly reduced the accumulation of QUIN at the site of injury and reduced the severity of delayed functional deficits in adult guinea pigs (Blight et al., 1995; Yates et al., 2006). This coincided with a significant 100% increase in the area of surviving white matter at the injury site in this model (Yates et al., 2006). A single dose of 4-Cl-3-HAA did not produce the same sensory and motor improvements suggesting that the benefits of continued 4-Cl-3-HAA treatment result from a reduction in QUIN rather than any early anti-oxidant activity (Yates et al., 2014).However, the use of 4-Cl-3-HAA did not significantly increase upstream neuroprotective kynurenic acid (KYNA) which may offer additional benefits in SCI. For example, a KYNA derivative (glucosamine-kynurenic acid) administered via spinal catheter resulted in improved motor function recovery at 1–4 weeks post-injury in male adult Wistar rats (Korimová et al.,2012) and KYNA administered by stereotaxic microinjection resulted in significant dose dependent improvements in hind limb function and reduced overall functional deficits following SCI (Wrathall et al., 1992). Taken together this suggests that targeting the KP and in particular KMO in order to shift the KP away from QUIN and towards KYNA may have several beneficial effects in SCI.
Another therapeutic benefit of targeting KMO in SCI was shown by treatment of spinal cord damaged rats with gene transfer of human KAT-II (the KP enzyme that leads to KYNA synthesis) which improved overactive bladder problems that can result from SCI (Jia et al., 2014). Mechanically,this improvement resulted from increased KYNA expression that antagonises the NMDA receptor. Previous studies have shown that supressing NMDA receptors improves detrusor activity that leads to bladder overactivity in SCI. Additionally, ketamine and other NMDA receptor antagonists have shown significant promise for the treatment of neuropathic pain, a common complication following SCI (Niesters and Dahan, 2012; Hagen and Rekand, 2015). In a rat model of neuropathic pain, induced following chronic constriction injury (CCI) of the right sciatic nerve, mRNA levels of IDO2, KMO and 3-HAO were significantly increased 7 days post-injury. Repeated administration of KMO inhibitors UPF-648 and Ro 61-8048 reduced hypersensitivity and neuropathic pain (Rojewska et al., 2016, 2018), most likely by shifting the KP towards the NMDA antagonist KYNA.
Conclusions and Future Directions
The data discussed above forms a conceptual framework that supports investigating the therapeutic potential of KMO inhibitors in SCI to not only reduce the pathophysiological impact of chronic inflammation, but to potentially improve psychological disturbances and neuropathic pain that can result from chronic SCI. Since the demonstration by Zwilling et al. (2011), that KMO inhibition ameliorated neurodegeneration and produced other positive results in mouse models of Alzheimer’s and Huntington’s diseases, much effort has focused on the development of KMO inhibitors.KMO is viewed as a better target than IDO-1, as KMO is present at thefirst junction of the KP and its inhibition leads to reduced QUIN but also increased neuroprotective KYNA.However, as pointed out by Zwilling et al. (2011), the therapeutic effects of the KMO inhibitor used did not result from reduced brain levels of QUIN but reflected the peripheral increase in KYN that is able to cross the blood-brain-barrier(BBB) and leads to increased brain KYNA (Zwilling et al.,2011). Newer, alternate, KMO inhibitors similarly fail to achieve good CNS exposure (CHDI-246 and UPF-648) and a good case exists for the development of a BBB-permeable KMO inhibitor that would achieve both therapeutic goals,i.e., reduced CNS QUIN with increased KYNA. In this vein,we recently commenced a KMO inhibitor discovery project.We utilized high-throughput screening (HTS), specifically using a lead-like structure library comprising a high proportion of leads likely to be CNS-permeable (Jacobs et al., 2018)and we hope to develop a BBB-permeable KMO inhibitor that could possibly be useful in SCI.
Acknowledgments:The authors thank Prof Gilles Guillemin, Macquarie University for helpful discussions.
Author contributions:Manuscript conception: DBL; literature search and manuscript writing: KRJ and DBL.
Conflicts of interest:None declared.
Financial support:None.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Hedong Li, Pennsylvania State University, USA.
Additionalfile:Open peer review report 1.
杂志排行

中国神经再生研究(英文版)的其它文章
- MicroRNAs of microglia: wrestling with central nervous system disease
- Huangqinflavonoid extraction for spinal cord injury in a rat model
- Apomorphine effects on the hippocampus
- Roles and functions of Atp6ap2 in the brain
- Magnesium sulfate and fetal neuroprotection:overview of clinical evidence
- Polyphenols-gut microbiota interplay and brain neuromodulation