液相色谱-串联质谱法同时测定中成药与保健食品中非法添加的9种尿酸调节类药物
2018-10-16张秋炎罗辉泰黄晓兰吴惠勤朱志鑫林晓珊谢梦婷吴庆晖马叶芬
张秋炎,罗辉泰,黄晓兰,吴惠勤,朱志鑫,黄 芳,林晓珊,谢梦婷,吴庆晖,马叶芬
(广东省测试分析研究所 广东省化学危害应急检测技术重点实验室 广东省中药质量安全工程技术研究中心,广东 广州 510070)
高尿酸血症是以体内嘌呤代谢紊乱、血尿酸增高为主要特征的病症[1],可诱发痛风、尿石症、慢性肾病等疾病[2],发病率在全球范围内呈上升趋势[3],并与肥胖、高血压、高血糖和血脂紊乱等代谢综合征相关[4]。高尿酸血症主要由尿酸生成过多或排泄不足所致,因此,目前临床上常用的尿酸调节类药物主要有两大类[5-8]:抑制尿酸生成的别嘌醇、奥昔嘌醇、非布司他、托匹司他等黄嘌呤氧化酶抑制剂,以及促进尿酸排泄的丙磺舒、磺吡酮、雷西纳德、阿卤芬酯等药物。此外,氯沙坦也可以促进尿酸排泄而作为尿酸调节类药物使用[9]。
近年来,市场上出现了宣称可以治疗高尿酸血症或缓解痛风症状的中成药和保健食品,而一些不法厂商为牟取暴利向其中非法添加上述尿酸调节类西药成分,以迅速增强疗效来吸引目标人群。在不知情的情况下长期使用会引起药物毒副反应,对消费者的身体健康造成严重威胁,甚至危及生命,如丙磺舒和磺吡酮等药物会引起造血功能障碍[10-11]。因此,建立中成药和保健食品中尿酸调节类药物的检测方法,对打击非法添加行为、保障消费者身体健康具有重要意义。
目前,针对中成药和保健食品中非法添加尿酸调节类药物的检测方法尚未见报道。本文通过优化样品前处理手段和色谱-质谱条件,首次建立了一次进样可同时测定中成药和保健食品中9种尿酸调节类药物的液相色谱-串联质谱法(LC-MS/MS),这些药物包括别嘌醇、奥昔嘌醇、非布司他、托匹司他、丙磺舒、磺吡酮、雷西纳德、阿卤芬酯和氯沙坦,化学结构式如图1。本法样品处理简便快速、成本低、灵敏度高、定性可靠、定量准确,已成功应用于日常分析检测,可为中成药和保健食品中非法添加尿酸调节类药物的定性筛查及定量分析提供技术支持,为政府相关部门提高监控水平提供科学依据和技术保障。

图1 9种尿酸调节类药物的化学结构式Fig.1 Chemical structures of nine uric acid regulation drugs
1 实验部分
1.1 仪器与试剂
Agilent 1200 SL Series RRLC/6410B Triple Quard MS快速高效液相色谱-串联四极杆质谱联用仪(美国Agilent公司);AS 3120超声波发生器(天津奥特赛恩斯仪器有限公司);赛多利斯TP-114电子天平(美国Sartorious公司);XW-80A快速混匀器(海门市麒麟医用仪器厂)。甲醇、乙腈(色谱纯,德国Merck公司);甲酸、乙酸、甲酸铵、乙酸铵(LC-MS级,美国Sigma公司);实验用水为二次蒸馏水,其余试剂均为分析纯。
9种标准品:奥昔嘌醇(Oxipurinol,纯度97%)、别嘌醇(Allopurinol,纯度98%)、雷西纳德(Lesinurad,纯度98%)和非布司他(Febuxostat,纯度98%)均购自加拿大TRC公司;托匹司他(Topiroxostat,纯度98.54%)购自美国MCE公司;氯沙坦(Losartan,纯度99.9%)和丙磺舒(Probenecid,纯度99.5%)购自中国食品药品检定研究院;磺吡酮(Sulfinpyrazone,纯度99%)和阿卤芬酯(Arhalofenate,简称MBX-102,纯度99%)购自美国Sigma公司。
所用的中成药和保健食品均为客户委托送检样品。
1.2 标准溶液的配制
分别准确称取各标准品10.0 mg(精确至0.01 mg)至10 mL容量瓶中,用甲醇溶解并定容至刻度(奥昔嘌醇、托匹司他不能完全溶于甲醇,需加少量二甲亚砜助溶),混匀得到1 000 mg/L的单标标准储备液,置棕色储液瓶中,于-20 ℃保存。临用时,以10%乙腈或空白基质提取液将上述标准溶液稀释成系列混合标准溶液。
1.3 样品前处理
固体样品:胶囊取内容物,片剂、丸剂、颗粒剂研磨成粉末后混匀。准确称取均匀试样1.0 g(精确至0.01 g)于10 mL具塞刻度试管中,加入2 mL水,涡旋1 min,加7 mL乙腈,涡旋混匀,超声提取20 min,取出,冷却至室温,用乙腈定容至刻度,混匀,准确移取1 mL至10 mL具塞刻度试管中,用水稀释至刻度,混匀,过0.22 μm滤膜后,供测定。
液体样品:充分摇匀。准确称取试样1.0 g(精确至0.01 g)于100 mL具塞刻度试管中,用10%乙腈定容至刻度,振摇混匀,过0.22 μm滤膜后,供测定。
1.4 液相色谱-串联质谱条件
1.4.1液相色谱条件色谱柱:Agilent Poroshell 120 Bonus-RP(100 mm×2.1 mm,2.7 μm,美国Agilent公司);流动相:A相为0.1%(体积分数,下同)乙酸溶液(含5 mmol/L甲酸铵),B相为乙腈;梯度洗脱程序:0~1.50 min,0%B;1.51~8.50 min,0%~50%B;8.50~8.51 min,50%~75%B;8.51~10.00 min,75%~95%B;10.00~11.50 min,95%B;11.50~11.51 min,95%~0%B;11.51~17.00 min,0%B。流速:0.4 mL/min;进样量:5 μL;柱温:30 ℃。
1.4.2质谱条件离子源:ESI;扫描模式:正离子;采集方式:多反应监测(MRM);干燥气(N2)温度:350 ℃;雾化气(N2)压力:276 kPa(40 psi);干燥气(N2)流量:9.0 L/min;毛细管入口端电压(Capillary):5 000 V;优化后的质谱采集参数见表1。

表1 9种尿酸调节类药物的分子式、保留时间及质谱采集参数Table 1 Molecular formula,retention time and MS parameters of nine uric acid regulation drugs
* quantitative ion
2 结果与讨论
2.1 质谱条件的确定
在电喷雾离子源的正离子和负离子模式下,分别对5 mg/L的9种待测物单标标准溶液作一级质谱全扫描分析。实验表明,9种尿酸调节类药物在两种电离模式下均有响应,正离子模式下可获得[M+H]+准分子离子峰,而负离子模式下可获得[M-H]-准分子离子峰。但在负离子模式下,奥昔嘌醇和别嘌醇的质谱响应优于正离子模式,且在负离子模式下为获得较高的质谱信号,需在流动相中加入氨水等碱性添加剂,而较高的pH值又限制了色谱柱的选择。综合考虑,选用正离子模式作进一步优化,通过选择离子监测(SIM)优化毛细管出口端电压(Fragmentor),获得各化合物的母离子响应最佳的碰撞电压,然后对这些母离子作子离子全扫描分析,根据欧盟2006/657/EC决议中有关质谱分析方法必须不少于4个识别点的规定[12],为各待测物选取质谱响应最佳的两个子离子作为特征碎片离子,通过MRM方式优化碰撞能量使其响应最佳。优化后的质谱参数见表1。最终根据色谱保留时间和两对MRM离子对的丰度比定性鉴别各组分,以MRM定量离子对的峰面积为依据进行定量测定。
2.2 色谱条件的优化
2.2.1色谱柱的选择9种尿酸调节类药物的极性跨度较大,由化学结构式可知,别嘌醇和奥昔嘌醇的极性偏大,在反相色谱上保留极弱,而其他7种待测物的极性较适中,在反相色谱中有较好的保留。因此,应侧重于选用对极性化合物有较好保留的反相色谱柱进行试验。试验比较了3款不同型号的色谱柱Poroshell 120 SB-C18(100 mm×2.1 mm,2.7 μm)(色谱柱A)、XSelect®HSS T3(100 mm×2.1 mm,3.5 μm)(色谱柱B)和Poroshell 120 Bonus-RP(100 mm×2.1 mm,2.7 μm)(色谱柱C)的分离效果,结果表明:色谱柱A对奥昔嘌醇以及别嘌醇的分离效果较优,但对丙磺舒、磺吡酮、阿卤芬酯、雷西纳德等的分离效果较差;色谱柱B和C均耐受100%水相环境,更适于分离极性化合物,但部分待测物在色谱柱B上的峰形拖尾。而色谱柱C对9种尿酸调节类药物尤其是别嘌醇和奥昔嘌醇均有较好的保留,且分离效果理想,峰形对称、峰宽较窄,所以选择Poroshell 120 Bonus-RP(100 mm×2.1 mm,2.7 μm)作为本法的色谱分离柱。
2.2.2流动相的选择流动相及其添加剂对待测物的色谱保留行为、峰形和质谱响应均有较大影响。在选定的色谱柱上,以乙腈为有机相,分别考察了0.1%甲酸、0.1%乙酸、5 mmol/L甲酸铵、5 mmol/L乙酸铵、0.1%乙酸(含5 mmol/L甲酸铵)、0.1%甲酸(含5 mmol/L甲酸铵)、0.1%甲酸(含5 mmol/L乙酸铵)和0.1%乙酸(含5 mmol/L乙酸铵) 8种常用水相对待测物的分离情况。结果显示,采用0.1%甲酸或0.1%乙酸作为水相时,质谱响应明显增强,但前者的分离度变差,而后者的保留时间延长、峰形展宽;采用5 mmol/L乙酸铵或5 mmol/L甲酸铵作为水相时,质谱响应明显减弱,化合物的分离效果变差。经反复试验,以0.1%乙酸(含5 mmol/L甲酸铵)为水相,乙腈为有机相,采用梯度洗脱,可以获得最佳分离效果,且待测物的色谱峰拖尾明显减少,但其质谱灵敏度受到较大影响。综合考虑,最终确定色谱分离条件见“1.4.1”,该条件下9种尿酸调节类药物均具有较好的质谱响应,良好的色谱保留和色谱峰形。图2为9种待测物混合标准溶液的MRM定量离子对色谱图。

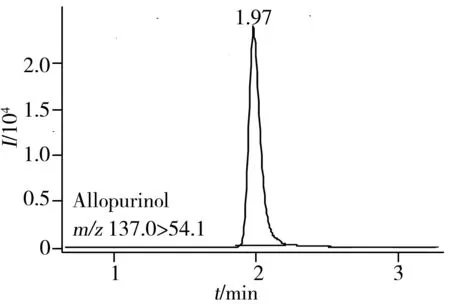





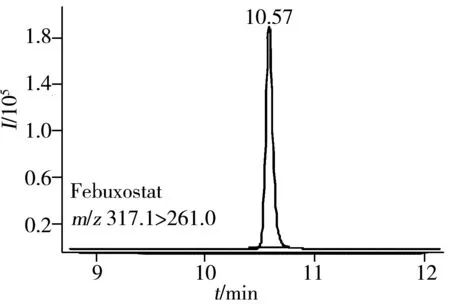
图2 9种调节尿酸类药物混合标准溶液的MRM定量离子对色谱图
Fig.2 MRM chromatograms of nine uric acid regulation drugs mixed standard solution
2.3 样品前处理条件的优化
提取溶剂的选择在样品预处理中显得尤为重要,而这又取决于目标化合物的理化性质和样品基质[13]。本研究涉及的9种尿酸调节类药物,结构和性质虽然差别较大,但在甲醇、乙腈中均具有一定的溶解度。中成药和保健食品多为酸性基质,且多含氨基酸和糖苷等,此类物质极性大,易溶于甲醇和水;而乙腈极性范围宽,对多数化合物均具有较好的溶解性和较高的提取率,对糖、脂肪、蛋白质化合物的提取率低,且分子小,组织穿透能力强[14]。因此,本研究分别考察了甲醇、水-甲醇、乙腈和水-乙腈作为提取溶剂的提取效果。结果表明,以甲醇、水-甲醇作为提取溶剂,易将极性较大的物质一并提取,造成较大的基质干扰;用纯乙腈作为提取溶剂时,部分待测物的提取回收率过低,而采用水-乙腈作为提取溶剂时,提取回收率明显提高,且基质影响不明显,但将该提取液直接进样,会影响奥昔嘌醇和别嘌醇等强极性待测物的色谱峰形,出现峰分叉或前延,这主要由于进样溶液的有机相比例过高所致。因此,实验选择水-乙腈为提取溶剂,采用先提取后用水稀释的方法进行样品处理,可以得到较理想的提取效果和色谱峰形。
2.4 基质效应的考察
与其他分析手段不同,质谱分析尤其是电喷雾质谱分析往往会存在基质效应(ME)[15-16],影响方法的灵敏度、精密度和准确度。本文采用提取后添加法对固体样品(胶囊)及液体样品(药酒)进行了基质效应评价,用10%乙腈溶液、阴性固体基质溶液及阴性液体基质溶液分别配制系列浓度的混合标准溶液,上机测定,考察各待测物的基质效应(ME)。采用公式ME=B/A(B为基质匹配标准曲线的斜率,A为纯溶剂配制标准曲线的斜率)计算ME,结果见表2。其中ME比值越接近1,说明基质效应越小,反之亦然。由表2可知,除雷西纳德的ME值偏低外,其他8种化合物ME值在0.86~1.11之间,呈弱基质效应,为消除或补偿基质效应给定量带来的偏差,本文采用配制空白基质匹配标准溶液的方法绘制标准曲线。
2.5 线性范围与检出限
在优化色谱-质谱条件下,对6个质量浓度水平的系列混合标准溶液进行测定,以各组分的MRM定量离子对峰面积(y)为纵坐标,对应的质量浓度(x,μg/L)为横坐标绘制标准曲线,各待测物在一定质量浓度范围内具有良好的线性关系,相关系数(r2)为0.995 7~0.999 9(见表2)。采用标准添加法测定,以定量离子对的信噪比(S/N)≥3确定待测物的检出限(LOD),S/N≥10确定待测物的定量下限(LOQ),得到9种化合物的LOD为0.03~0.6 mg/kg,LOQ为0.1~2.0 mg/kg,详见表2。

表2 9种尿酸调节类药物的线性方程、相关系数、线性范围、检出限、定量下限及基质效应Table 2 Linear equations,correlation coefficients(r2),linear ranges,LODs,LOQs and matrix effects of nine uric acid regulation drugs
y:peak area;x:mass concentration,μg/L
2.6 回收率与相对标准偏差
取阴性固体样品(胶囊)及阴性液体样品(药酒),在低、中、高3个加标水平下分别进行加标回收试验,每个加标水平按“1.3”方法处理后平行测定6次,计算平均回收率和相对标准偏差(见表3)。结果显示,方法的平均回收率为79.2%~108%,相对标准偏差(RSD,n=6)为1.2%~12%,具有良好的准确度和精密度,能满足9种尿酸调节类药物的测定要求。
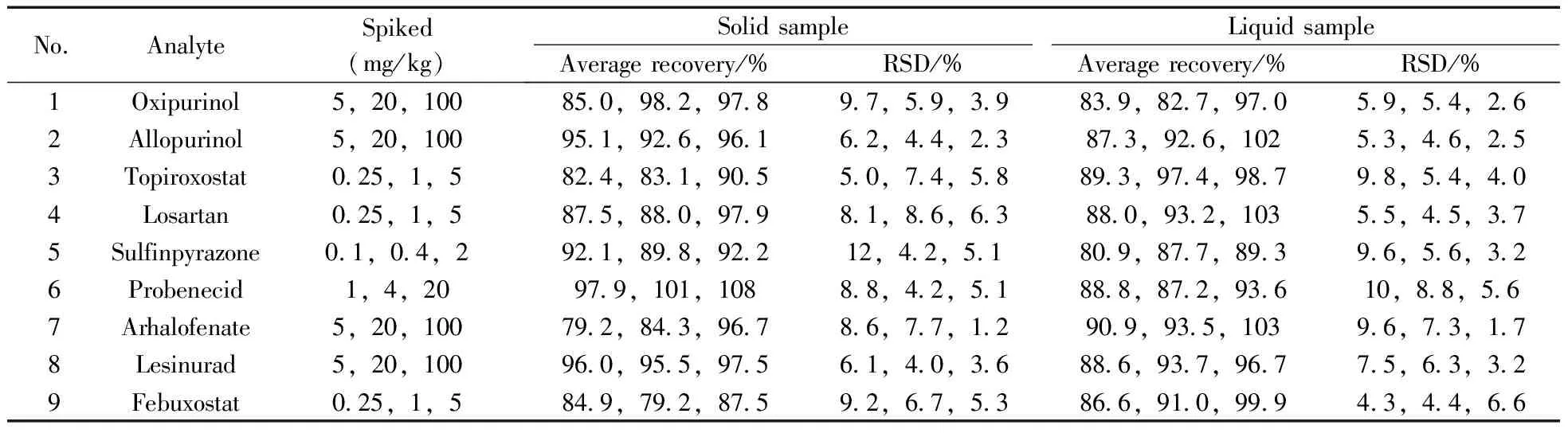
表3 9种尿酸调节类药物的回收率及相对标准偏差(n=6)Table 3 Average recoveries and RSDs of nine uric acid regulation drugs(n=6)
2.7 实际样品的测定
采用所建方法对客户委托送检的12批次中成药或保健食品(其中胶囊3批次、片剂1批次、颗粒5批次、丸剂2批次和药酒1批次)进行测定,均未检出本文涉及的9种尿酸调节类药物。
3 结 论
本研究首次建立了LC-MS/MS同时快速测定中成药和保健食品中9种非法添加尿酸调节类药物的新方法。对样品处理手段、色谱分离条件以及质谱采集参数进行了优化。方法操作简单、快速、溶剂用量少、灵敏度高、结果准确可靠,可为宣称治疗高尿酸血症或具有抗痛风功效的中成药和保健食品中非法添加化学药物的监测提供技术支持。
