臭柏叶绿体基因组结构与系统进化分析
2018-10-16路东晔郭文雨李娅翔张玉琨张国盛
路东晔,张 磊,郝 蕾,郭文雨,李娅翔,张玉琨,张国盛
(内蒙古农业大学 林学院,呼和浩特 010019)
叶绿体是一种绿色植物发生光合作用和能量转化的重要细胞器,起源于内共生蓝藻[1]。叶绿体基因组(cpDNA)一般是双链环状。大多数植物叶绿体基因组是母性遗传,而裸子植物中为父系遗传[2]。叶绿体基因组与核基因组和线粒体基因组相比,不仅具有基因组较小、四分体的保守结构、易于测序,单亲遗传无基因污染,转基因表达稳定、环境安全性高、无基因沉默的特征[3],而且叶绿体基因组的核苷酸进化速率适中,在编码区(CDS)和非编码(CNS)区之间差异明显。这导致叶绿体基因组适用于不同分类学水平的系统发育研究。在DNA条形码的开发、物种亲缘关系鉴别、叶绿体基因工程、植物药材真伪鉴定、食品质量控制、出入境植物检验检疫等方面得到广泛应用[4-5]。自从1986 年人类首次获得烟草(Nicotianatabacum)[6]和地钱(Marchantiapolymorpha)[7]叶绿体基因组以来,截止到2016年10月 31日,发现目前针对叶绿体基因组的研究涉及200 个科属,叶绿体基因组的长度集中在 140~160 kb,GC 含量多为 35%~40%,编码 80~100 个基因[8]。截至2017年3月,NCBI中已经公布的陆生植物叶绿体基因组数据就达到1 157条[9]。这些研究表明针对叶绿体基因组的研究呈逐年上升的趋势,利用这些叶绿体基因组序列精确的揭示植物系统发育关系,已成为分析植物系统发育和群体遗传学最有效的途径。
植物的核基因组较复杂,基因操作比较困难,时间、资金成本较高,且很难筛选得到有效的基因进行系统进化分析,相反叶绿体基因组因操作简单、成本低等优势,被广泛应用。随着柏科植物叶绿体基因组的陆续公布,一些问题随之产生。首先,这些柏科植物基因组的结构是否存在差异?基于叶绿体基因组序列数据,柏科植物间的系统发育关系是怎样的?臭柏(Juniperussabina)又名叉子圆柏、沙地柏、新疆圆柏等,属柏科刺柏属。常绿匍匐针叶灌木,成岛屿状天然分布于欧洲、中亚及中国西北天山、祁连山、内蒙古贺兰山、阴山、青海东北部等干旱贫瘠环境中,具有适应性强、耐旱性强的特点,根系发达,生长旺盛,不仅有保持水土、防风固沙、造林绿化的生态价值[10],而且还有经济价值[11]和医学价值[12]。臭柏分布范围如此之广,是研究进化历史的优良材料,因单亲遗传容易受到群体遗传效应的影响,故而叶绿体分子标记可产生更清晰的历史踪迹。本研究采用高通量测序技术进行臭柏叶绿体全基因组测序,通过组装、拼接得到叶绿体基因组全长,并对其组成、结构及系统发育进行分析,旨在丰富臭柏的遗传信息,为未来研究臭柏的谱系地理和柏科刺柏属植物间的系统进化及亲缘关系提供理论依据。
1 材料和方法
1.1 实验材料
选取位于毛乌素沙地鄂尔多斯市乌审旗沙地柏自然保护区境内天然分布、长势较好的臭柏植株(N38°23′,E108°44′),采集其新鲜叶片作为实验材料,经锡纸包裹、液氮速冻后存于-80 ℃保存备用。
1.2 研究方法
1.2.1全基因组DNA的提取及测序采用植物基因组DNA提取试剂盒(TIANGEN)提取臭柏全基因组DNA,并送样测序。样品由上海美吉生物有限公司质检合格后,采用Illumina Hiseq X Ten双端测序技术,测序读长为PE150,构建文库大小为460 bp。
1.2.2叶绿体基因组组装与特征分析构建好文库后,通过桥式PCR获得测序数据,由于Illumina Hiseq 的原始测序数据会存在一些质量比较低的数据,为了使后续的组装更加准确,会对原始数据进行质量剪切,将质量较低的数据作如下处理:1) 去除reads 中的adapter 序列;2) 剪切前去除5′端含有非A、G、C、T 的碱基;3) 修剪测序质量较低的reads 末端(测序质量值小于Q20);4) 去除含N 的比例达到10%的reads;5) 舍弃去adapter 及质量修剪后长度小于25 bp 的小片段。对质量剪切后的数据利用SOAPdenovo v2.04(http://soap.genomics.org.c n/)[13]拼接软件对优化序列进行多个Kmer参数的调整,得到最优的组装结果。运用GapCloser v1.12 软件对组装结果进行局部内洞填充和碱基校正。将组装结果放入NCBI数据库中比对找到相似度最高的参考基因组(Juniperusvirginiana),利用DOGMA 软件(http://dogma.ccbb.utexas.edu/)[14]对基因组中包含的蛋白编码基因、rRNA 和tRNA 进行预测,然后根据参考基因组进行人工矫正。通过OGDRAW v1.2(http://ogdraw.mpimp-golm. mpg.de/cgi-bin/ogdraw.pl)[15]软件对注释结果进行可视化,绘制叶绿体基因组物理图谱。利用Sequin软件完成臭柏叶绿体基因组的提交。利用CodonW[16]软件分析蛋白编码基因密码子偏好性。通过RepeatMasker (http://www. repeatmasker.org/cgi-bin/WEBRepeatMasker) 软件进行散在重复序列预测,TRF(Tandem Repeats Finder) (http://tandem.bu.edu/trf/trf.submit.options.html)搜寻DNA 序列中的串联重复序列,MISA(Microsatellite Identification Tool)软件(http://pgrc.ipk-gatersleben.de/misa/misa.html)预测微卫星重复序列,参数设置为单元大小unit size,最小重复数min repeats: 1~10、2~6、3~5、4~5、5~5、6~5; 2个SSR 之间的最小距离设置为100 bp。参数意义即1 个碱基重复10 次及以上;2 个碱基重复6 次及以上; 3个碱基、4 个碱基、5 个碱基和6个碱基分别重复5 次及以上。同时,2个微卫星之间的距离小于100 bp 时,2个微卫星组成1个复合微卫星。
1.2.3基于叶绿体基因组序列系统进化分析首先用MAFFT[17]软件对NCBI数据库已公开发表的柏科刺柏属Juniperuscedrus、Juniperusvirginiana、Juniperusbermudiana、欧洲刺柏(Juniperuscommunis)、刺柏(Juniperusformosana)、Juniperusmonosperma和Juniperusscopulorum等7种植物连同臭柏以及柏科、杨柳科和蔷薇科部分植物共31种进行序列比对,后采用RAxML[18]软件构建系统发育树,利用最大似然法(ML)对数据进行分析。
2 结果与分析
2.1 臭柏叶绿体全基因组结构及特征
臭柏叶绿体基因组测序得到10 G左右的原始数据,其中包含2 542 965个片段(reads),标准片段长度为150 bp,通过去掉低质量序列后得到2 424 078条reads用于组装,平均测序深度达到752个碱基,经检验合格后,将臭柏叶绿体基因组序列提交到GenBank数据库,登录号为MH324744。
组装好的叶绿体基因组全长127 739 bp,GC总含量为34.9%,其中碱基A占33.1%、碱基C占16.9% 、碱基G占 18.0%、碱基T占 32.0%,具有叶绿体典型的四分体结构。其中大单拷贝区(large single copy, LSC)长91 264 bp,小单拷贝区(small single copy, SSC)长35 952 bp,两者被两段长度相等为261 bp反向重复区(inverted repeat region) IRa和IRb分隔(图1)。臭柏叶绿体基因组包含119个基因。其中包括82个蛋白编码基因、4个rRNA基因和33个tRNA基因。其中trnI-CAU和trnQ-UUG基因有2个拷贝,其他基因均为单拷贝基因。多拷贝基因中只有trnQ-UUG位于IR区。CDS全长75 498 bp,占叶绿体基因组全长的59.1%,GC含量为36.3%,编码25 166个氨基酸;基因间区总长度52 241 bp,占基因组全长的40.9%,GC含量为32.9%;在臭柏叶绿体基因组中注释到有14个基因含有内含子,其中有12个基因含有1个内含子,2个基因(rps12和ycf3)含有2个内含子。最大内含子为trnI-GAU基因所有,长844 bp,相反,trnL-UAA基因包含长288 bp最小内含子(表1)。

表1 臭柏叶绿体基因组包含外显子和内含子的基因
注:rps12为反式剪接基因
Note: Therps12 is a trans-splicing gene
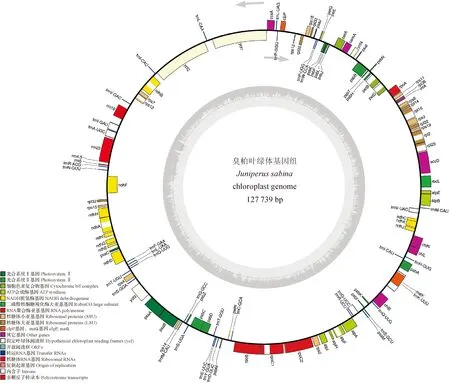
图1 臭柏叶绿体基因组物理图谱Fig.1 The gene map of chloroplast genome in J. sabina
按照功能臭柏叶绿体的基因可分为三类,包括转录和翻译相关基因、光合作用有关基因以及与脂肪酸和氨基酸等合成有关基因(表2)。
2.2 臭柏叶绿体基因组密码子偏好性
使用Codon W软件分析臭柏叶绿体基因密码子偏好性,发现在臭柏叶绿体基因组共有42 579个密码子,其中编码亮氨酸Leu的数量最多,有2 750个,占总数的6.46%;编码半胱氨酸Cys的数量最少,有293个,占密码子总数的0.69%。相对同义密码子使用度最高的为AGA/UUA,最低的是CGG。在密码子使用度(RSCU)大于1的氨基酸中,除trnL-CAA对应密码子是G结尾外,其他均以A/U结尾(图2)。
2.3 臭柏叶绿体基因组重复序列
重复序列可分为串联重复序列(tandam repeat)和散在重复序列(interpersed repeat)两大类。通过软件预测在臭柏叶绿体基因组中存在49个串联重复序列,长4 594 bp,占基因组总长的3.6%。其中长度最小为4 bp,最大为99 bp,20 bp~30 bp的重复序列最多,有26个。在臭柏叶绿体基因组中共预测到47个SSR,包括38个单核苷酸,1个二核苷酸SSR,3个三核苷酸SSR,5个复合型SSR。78.7%的SSR位于IGS和内含子等非编码区域且A/T碱基在SSR中出现频率较高,在38个单核苷酸SSR中,包含A碱基SSR有20个,包含T碱基SSR有18个(表3)。从表中可以看出SSR在叶绿体基因组中分布不均匀,可为未来研究臭柏种群遗传多样性和遗传结构提供参考。没有预测到散在重复序列。
2.4 刺柏属植物种间叶绿体基因组比较
将臭柏叶绿体基因组与NCBI数据库中已提交的其他刺柏属的植物叶绿体基因组进行比较,发现刺柏属8个种间的叶绿体基因组大小、反向重复区大小及GC含量相近,基因数目和类型相同(表4)。
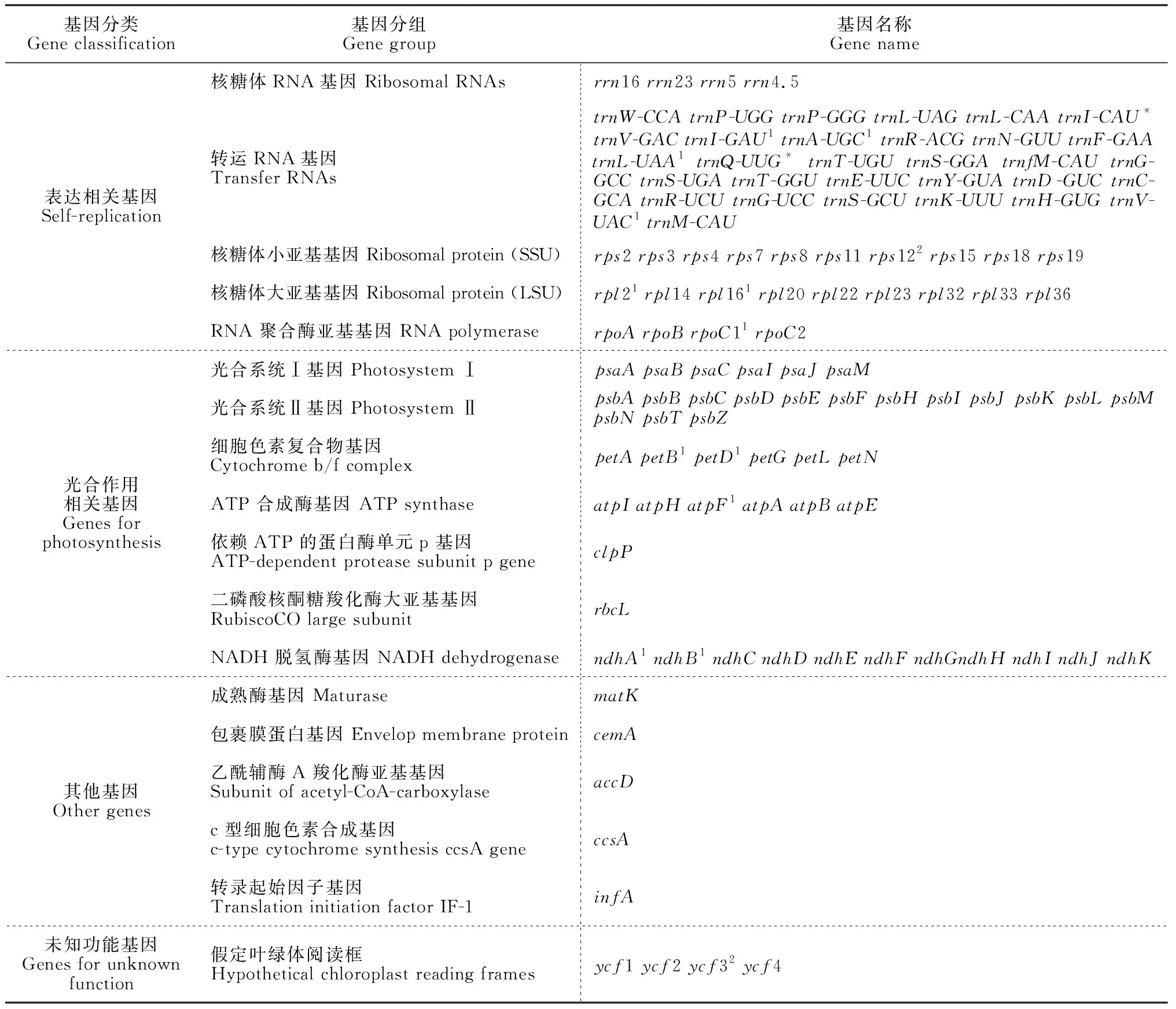
表2 臭柏叶绿体基因组注释基因分类表
注:上标*、1、2分别表示多拷贝基因、含有1个内含子基因和含有2个内含子基因
Note: *, 1 and 2 represent multiple copies of the gene, contain one intron of the gene, and of the gene contain two introns respectively
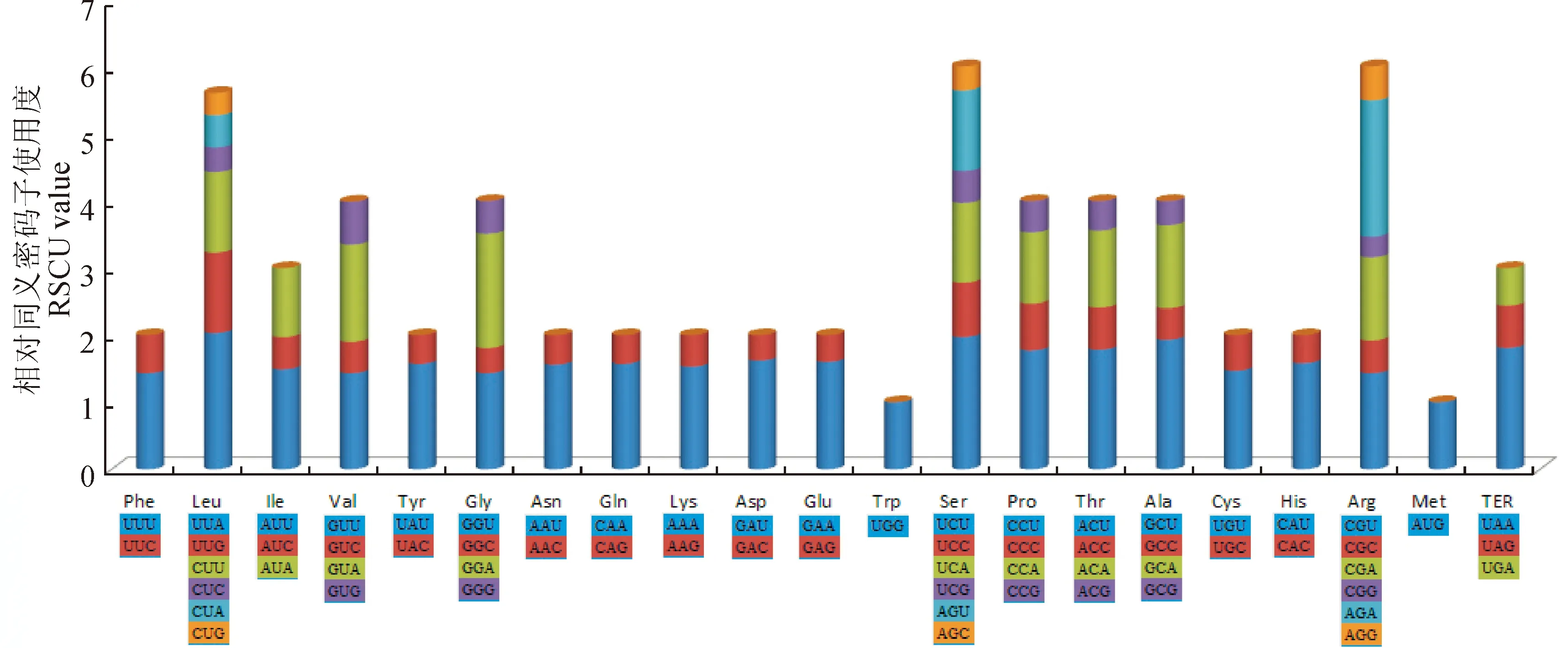
图2 臭柏叶绿体基因组氨基酸相对同义密码子使用度RSCUFig.2 The RSCU of codons in J.sabina chloroplast genome
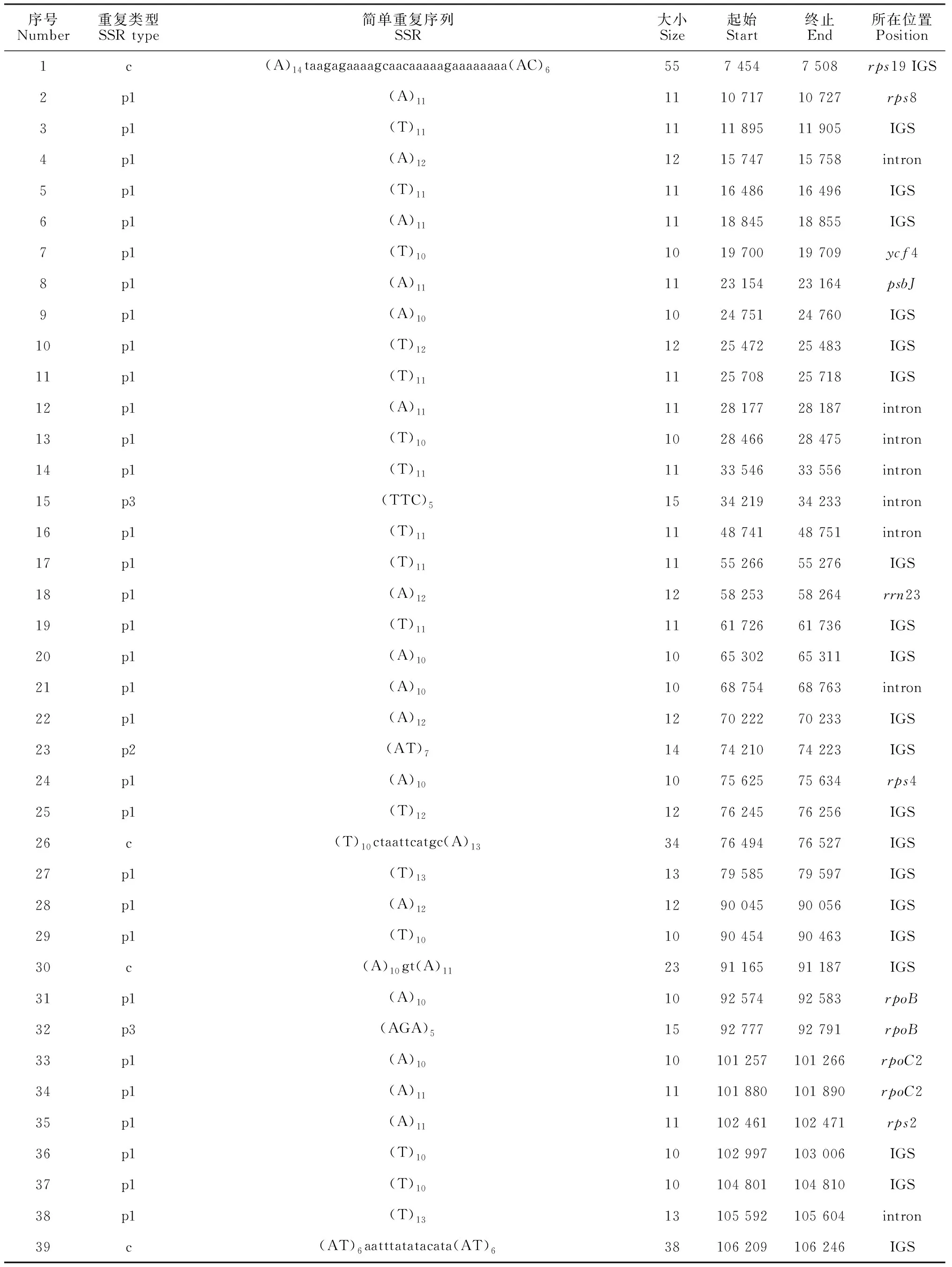
序号Number重复类型SSR type简单重复序列SSR大小Size起始Start终止End所在位置Position1c(A)14taagagaaaagcaacaaaaagaaaaaaaa(AC)6557 4547 508rps19 IGS2p1(A)111110 71710 727rps83p1(T)111111 89511 905IGS4p1(A)121215 74715 758intron5p1(T)111116 48616 496IGS6p1(A)111118 84518 855IGS7p1(T)101019 70019 709ycf48p1(A)111123 15423 164psbJ9p1(A)101024 75124 760IGS10p1(T)121225 47225 483IGS11p1(T)111125 70825 718IGS12p1(A)111128 17728 187intron13p1(T)101028 46628 475intron14p1(T)111133 54633 556intron15p3(TTC)51534 21934 233intron16p1(T)111148 74148 751intron17p1(T)111155 26655 276IGS18p1(A)121258 25358 264rrn2319p1(T)111161 72661 736IGS20p1(A)101065 30265 311IGS21p1(A)101068 75468 763intron22p1(A)121270 22270 233IGS23p2(AT)71474 21074 223IGS24p1(A)101075 62575 634rps425p1(T)121276 24576 256IGS26c(T)10ctaattcatgc(A)133476 49476 527IGS27p1(T)131379 58579 597IGS28p1(A)121290 04590 056IGS29p1(T)101090 45490 463IGS30c(A)10gt(A)112391 16591 187IGS31p1(A)101092 57492 583rpoB32p3(AGA)51592 77792 791rpoB33p1(A)1010101 257101 266rpoC234p1(A)1111101 880101 890rpoC235p1(A)1111102 461102 471rps236p1(T)1010102 997103 006IGS37p1(T)1010104 801104 810IGS38p1(T)1313105 592105 604intron39c(AT)6aatttatatacata(AT)638106 209106 246IGS
续表3 Continued Table 3

序号Number重复类型SSR type简单重复序列SSR大小Size起始Start终止End所在位置Position40p1(A)1414108 280108 293intron41p1(T)1111113 116113 126IGS42p3(AGA)618113 564113 581IGS43p1(T)1010113 829113 838IGS44p1(A)1111114 136114 146intron45c(A)11ttgaccggattcaatttattgtgattttctataatgaatcaaatgatgagtggttgacgatataatttgtg(TA)796118 513118 608IGS46p1(A)1717123 740123 756IGS47p1(A)1212126 908126 919IGS
注:p单个SSR类型;p1/p2/p3中数字分别表示构成基序的碱基个数;c复合SSR类型
Note: p indicates single SSR type; the numbers in p1/p2/p3 indicate the number of bases constituting the motif, respectively; c indicates composite SSR type
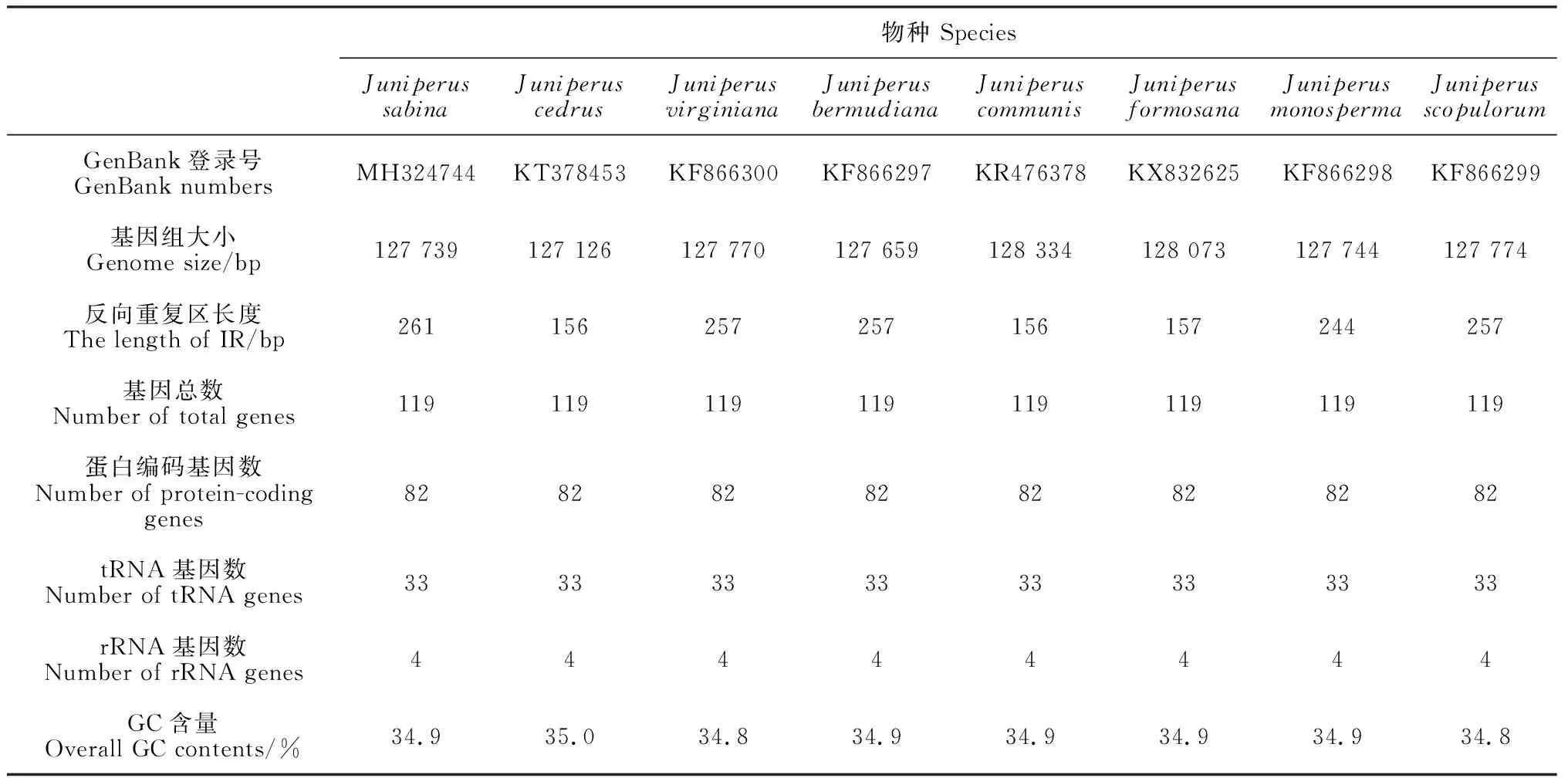
表4 刺柏属植物叶绿体基因组比较
叶绿体基因组最大的是J.communis,长128 334 bp,最小的为J.cedrus,长127 126 bp,两者相差1 208 bp。总的来说,刺柏属植物在进化中结构还是相对保守的。本研究中刺柏属植物的反向重复区大小为156~261 bp,这与柏科植物中日本柳杉(Cryptomeriajaponica)和Taiwaniacryptomerioides的IR只有大约280 bp且包含trnQ-UUG基因的研究结果相似[19],但重复拷贝基因trnQ-UUG间基因的排列形式不同,经过比对,臭柏与J.virginiana[20]的排列顺序相同(图3)。
以刺柏属植物Juniperuscedrus为参考基因组,通过mVISTA软件将臭柏连同刺柏属其他植物进行比较分析,发现非编码区变异较编码区大,差异大的区域可能为将来的研究提供候选DNA条形码。编码区中除accD、clpP、psbT、ycf1和ycf2等基因变异较大外,其他基因相对保守(图4)。
2.5 系统进化分析
以毛果杨、油松等7种植物作为外类群,连同NCBI数据库中23种柏科植物采用RAxML软件ML法(maximum likelihood,最大似然法)构建系统进化树。结果表明,进化树整体符合形态学分类,被子植物和裸子植物整体聚为两大类。被子植物分为蔷薇科和杨柳科两支,裸子植物分为松科和广义柏科[21]两支。广义柏科中所有刺柏属植物聚为一类。
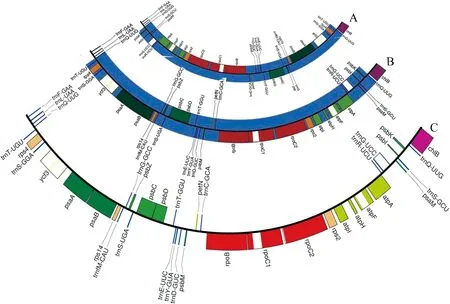
A物种为Juniperus scopulorum;B物种为Juniperus virginiana;C物种为臭柏图3 Juniperus scopulorum, Juniperus virginiana和臭柏trnQ-UUG基因间区结构排列A is Juniperus scopulorum, B and C represent Juniperus virginiana and Juniperus sabina, respectivelyFig.3 The intergenic structure arrangements of trnQ-UUG gene among Juniperus scopulorum,Juniperus virginiana and Juniperus sabina
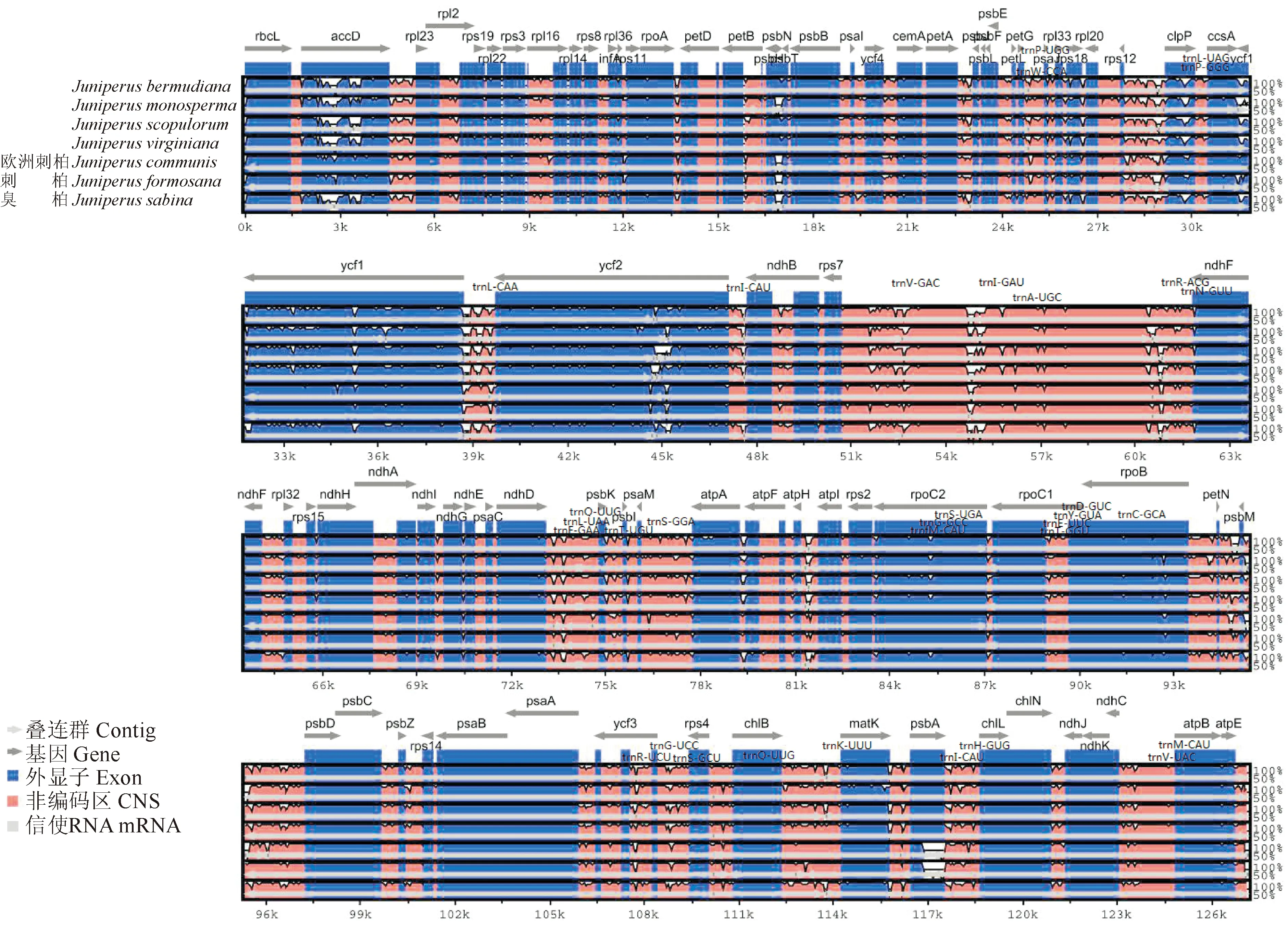
图中箭头表示基因方向,横轴表示基因组大小,纵轴表示50%~100%的相似度,不同颜色代表基因组中不同区域图4 刺柏属植物叶绿体基因组结构比较Arrows in the figure indicate gene orientation, horizontal axis indicates genome size, and vertical axis indicates 50% to 100% similarity. Different colors indicate different regions in the genomeFig.4 Comparison of chloroplast genomic structures in Juniperus plants
从进化树中(图5)可以看出,臭柏与J.bermudiana亲缘关系相对较近,而刺柏属植物整支为单系类群且与Cupressus,Hesperocyparis-Callitropsis进化枝的亲缘关系最近,这与Mao 在2010年基于叶绿体基因片段和核基因转录间隔区序列构建的进化树研究结果一致[22]。
中国植物志中将柏科分为翠柏属(Calocedrus)、扁柏属 (Chamaecyparis)、柏木属(Cupressus)、福建柏属(Fokienia)、刺柏属(Juniperus)、侧柏属(Platycladus)、崖柏属(Thuja)和罗汉柏属(Thujopsis)8个属[23],本研究中除福建柏属外均有涉及,本研究聚类结果也与植物志中分类基本相同(图5虚线方框内)。
3 讨 论
植物叶绿体基因组长度一般为107~218 kb,由小单拷贝区(SSC,长约18~20 kb)、大单拷贝区(LSC,长约 81~90 kb)以及2个反向重复区域 (IR,长约20~30 kb)[24]四部分组成。大多数植物叶绿体基因组结构保守,但基因组大小却不尽相同,如银杉(Cathayaargyrophylla)叶绿体基因组大小为107 kb[25],而天竺葵(Pelargoniumhortorum)却高达218 kb[26]。叶绿体基因组差异主要是由反向重复区的收缩、扩张或缺失引起的,而IR在稳定叶绿体基因组结构[27]和影响叶绿体基因组大小方面起着非常重要的作用[28]。纤细裸藻(Euglenagracilis)含有3个方向相同的串联重复DNA[29],天竺葵属(Pelargonium)反向重复区长75.7 kb[26],日本柳杉的反向重复区仅为114 bp[30],而牻牛儿苗属(Erodium)[31]和一些豆科植物[32-33]则无反向重复区,后有学者在蝶形花科[34]以及松、柏科中部分植物[35]中均发现了IR区全部或部分缺失。
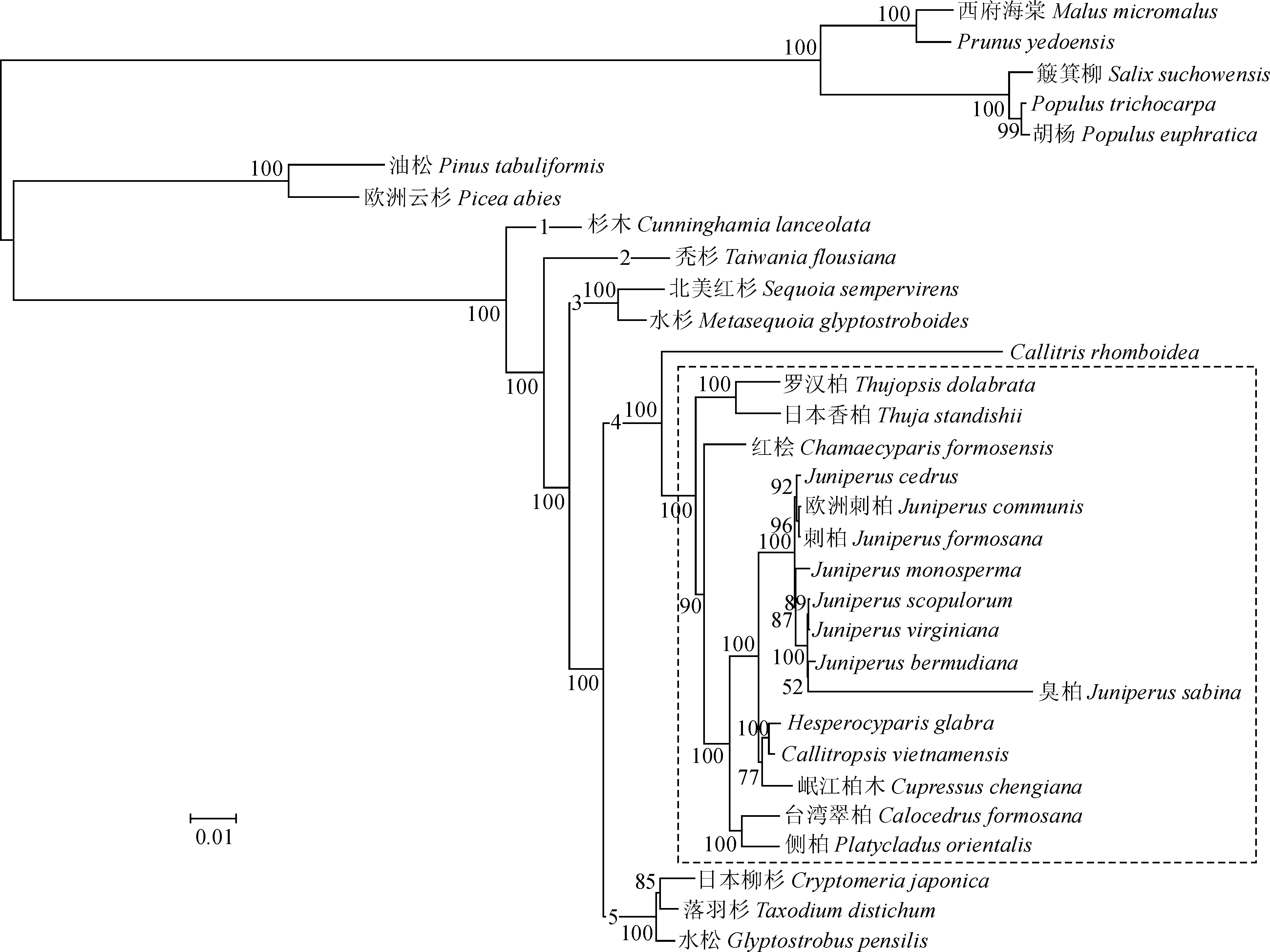
1杉木亚科;2台湾杉亚科;3红杉亚科;4澳洲柏亚科;5柏木亚科;6落羽杉亚科图5 基于最大似然法构建的31种植物聚类结果1-6 represent Cunninghamioideae, Taiwanioideae, Sequoioideae, Callitroideae, Cupressoideae, Taxodioideae respectivelyFig.5 Clustering results of 31 plants based on maximum likelihood
本研究对臭柏叶绿体基因组研究后发现,臭柏反向重复区与其他几种柏科植物具有相同特征:IR区只有大约156~261 bp且包含trnQ-UUG基因。在前人的研究中发现IR序列可促进高频同源重组以产生异构基因组形式。在刺柏属植物小单拷贝区中存在不同的异构形式“A”排列(J.monosperma和J.virginiana)和“B”排列(J.bermudiana和J.scopulorum)(以trnQ-UUG为界),从而表明这种大约250 bp的trnQ-IR已经在刺柏属中具有促进同源重组的活性[20]。臭柏作为刺柏属一员,其IR与“A”排列一致,表明可能也具有促进同源重组活性。有学者对松柏等针叶树研究后发现柏科植物缺失了IRB 区,而松科植物缺失IRA区[36],但因研究对象有限还没有普遍性的结论,值得肯定的是由于IR区的可变性可以作为研究植物进化事件的重要标志。
前人关于柏科分类有不同的见解。Gadek等基于matK和rbcL等2个叶绿体片段[37]将广义柏科划分为7个亚科,即杉木亚科(Cunninghamioideae)、台湾杉亚科(Taiwanioideae)、密枝杉亚科(Athrotaxidoideae)、红杉亚科(Sequoioideae)、落羽杉亚科(Taxodioideae)、澳洲柏亚科(Callitroideae)和柏木亚科(Cupressoideae)。而Fajorn[38]于2005年基于形态学证据提出最新的柏科分类系统,将柏科分为杉木亚科、台湾杉亚科、密枝杉亚科、红杉亚科、落羽杉亚科和柏木亚科6个亚科,将澳洲柏亚科降级并入柏木亚科。本研究中除密枝杉亚科外其他均有涉及,进化树与Gadek和Fajorn分类一致,且进化树中Juniperus,Cupressus和Hesperocyparis-Callitropsis聚为一类,这与Little[39]和QU[40]的研究结果基本一致。Little将新大陆柏木和越南柏属合并为广义卡利卓普西斯柏属(Callitropsissensu lato),此外,翠柏属(Calocedrus)和侧柏属(Platycladus)聚为一支,福建柏(Fokienia)嵌套在扁柏属(Chamaecyparis)内形成一支,罗汉柏属(Thujopsis)和崖柏属(Thuja)聚为一支。本研究中因没有福建柏数据,难以判断福建柏与扁柏关系,但其他属间关系基本相似,从而进一步验证了柏科的分类系统。臭柏自举值为52,较其他植物偏低,且枝长较长,进化速率快,说明臭柏与本研究中其他属内植物存在显著差异,这可能是由于形态差异与生长环境不同所导致。本研究涉及到的刺柏属植物中,除臭柏为灌木外,其他均为乔木;除刺柏产于中国台湾、西藏和浙江外,其他均原产自北美洲和欧洲[41]。此外,有研究表明臭柏属于圆柏属[41],这也为臭柏与刺柏属植物关系较远的结果提供了依据,但现今还没有圆柏叶绿体基因组序列,很难断定它们的关系,后续应着手圆柏叶绿体基因组测序工作,来进一步验证它们的关系。
多年来,不少学者大多通过叶绿体和核基因片段重建了部分柏科植物的系统发生关系[39,42-43]。如Xiang等基于核基因片段提出越南柏属和刺柏属都起源于柏木属内[44],也有学者对翠柏属[45]和刺柏属[46]等少数属内和属间关系作了研究,但是大部分属间关系以及属内的系统发育关系仍有待进一步完善,本研究首次采用高通量测序技术完成了臭柏叶绿体基因组序列、结构和系统进化分析,虽然丰富了柏科的遗传资源,但因数据库中提交的叶绿体基因组序列有限,基于叶绿体基因组序列构建柏科植物的系统进化关系还不够全面,未来将需要利用更多的数据来挖掘柏科的系统进化关系。
