Y分子筛催化异丁烷/丁烯烷基化反应中焦组分的吸附模拟
2018-10-10杜延年郭锦标
杜延年, 周 祥, 周 涵, 郭锦标
(中国石化 石油化工科学研究院, 北京 100083)
分子筛作为催化剂以其规则有序的孔道结构、良好的水热稳定性、可调控的酸强度、较高的初始活性和择形选择性而被广泛应用于石油炼制工业[1-3],随着汽油质量标准的不断升级,对环境更为友好的分子筛催化异丁烷/丁烯烷基化反应生产烷基化油技术被广泛研究,但在反应过程中分子筛易生焦,寿命较短。为解决烷基化反应过程中分子筛的失活问题,科研人员做了大量研究,意图解释分子筛的失活机理,目前主要有3种说法[4-7]。第一,直接失活模式,反应过程中生成的焦组分不可逆地吸附在酸中心,致使分子筛酸中心数量衰减而失活;第二,间接失活模式,反应过程中生成的焦类组分尺寸较大,孔道内扩散速率较慢,且空间位阻效应明显,易切断反应物分子向酸中心的扩散路径而导致失活;第三,两种失活模式并存。但以上说法仍存在以下疑点:第一,焦组分的表征手段受限,反应过程中生成的焦类物质的定性和定量分析不全面,致使反应过程的生焦机理研究并不明确;第二,不同结构类型的焦组分对分子筛性能的影响无法通过实验进行有效界定和区分,而现有的说法多是基于实验现象的宏观推测,尚未形成共识性结论。现有关于分子筛催化烷基化反应过程中的焦组分对分子筛性能影响的定性尚未见报道。笔者采用分子模拟方法,从微观层面研究烷基化反应过程生成的焦组分在Y分子筛孔道内Brönsted(B)酸中心附近的吸附过程,计算稳定吸附构象及相关参数,对比分析不同结构类型焦组分在分子筛孔道内的吸附差异性及对孔道空间的占据情况,为进一步研究分子筛的失活机理提供理论依据。
1 Y分子筛簇模型的建立与分子模拟方法
1.1 模型化合物的选择
Y分子筛催化异丁烷/丁烯烷基化反应过程生成的焦组分结构类型复杂多样,根据文献[4,6,8,10-12]报道的焦组分定性分析结果,对焦组分的结构特点进行总结:(1)焦组分碳数分布较广,一般在12~35之间,但多集中在15~20之间;(2)焦组分结构类型多样,多由直链烷烃类、直链烯烃类、环烷烃类和芳烃类及部分大分子正碳离子组成,高度支链化,环状结构多为单环或双环。结合烷基化反应机理和焦组分的结构信息,笔者以大分子直链烃裂解生成的C7正碳离子为直链类焦组分前驱体,C7烯烃类正碳离子为环状类焦组分前驱体,选取碳数为11、15和19的烷烃类(ALK)、烯烃类(OLE)、烷基环己烷类(ACH)、烷基萘烷类(ADE)、烷基苯类(ALB)和烷基萘类(ANA)等焦组分为模型化合物。
1.2 Y分子筛簇模型的建立
Y分子筛超笼由方钠石笼和六方柱笼连接而成,笔者截取300T Y分子筛簇模型,如图1所示。该簇模型由8个超笼组成,超笼之间通过共用十二元环连接,模型的边界用氢原子饱和,形成Si—H键,键长固定为0.147 nm,B酸中心选取O1位。采用量子力学/分子力学组合的方法计算时,图1(b)中绿色区域为量化区域,48T,处于弛豫状态,剩余部分原子为力学区域,被固定在其晶体结构上。
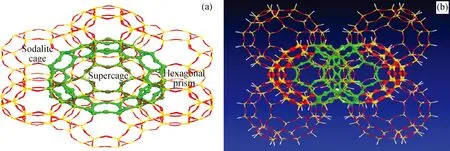
图1 Y分子筛模型及8个超笼的簇模型示意Fig.1 Schematic diagram of Y zeolite model and cluster model of 8 supercages (a) Y zeolite model ; (b) Cluster model of 8 supercages
1.3 计算方法和参数设置
计算流程大致如下,首先判定焦组分的吸附位原子,将焦组分的吸附原子设置在酸中心附近合适位置,采用Forcite模块中的模拟退火方法计算得到体系的全局最低能量吸附构象,然后采用Forcite模块中分子力学方法对吸附构象进行结构优化,最后通过量子力学/分子力学(QM/MM)组合方法的QMERA模块对焦分子和分子筛量化区域的空间结构和电子性质做进一步优化得到稳定吸附构象。
采用BIOVIA公司的Materials Studio8.0软件中基于QM/MM组合方法的Qmera模块,其中QM计算时,选用广义梯度近似GGA的PBE泛函,DNP基组,自洽场(SCF)迭代过程当中能量、受力和位移的收敛阈值分别为2.62×10-2kJ/mol、0.5251 kJ/(mol·nm)和0.0005 nm。MM计算时,采用Universal力场[13],Additive方式计算体系能量,计算精度选择Fine。Forcite模块进行退火计算时,精度选择Fine,力场为Universial,静电力和范德华力模拟方法均选择Atom based。
采用公式(1)~(4)计算焦组分在分子筛孔道内吸附时的吸附能、范德华作用能、与B酸中心的相互作用能。
Eads=EZ-OH-coke-EZ-OH-Ecoke
(1)
(2)
(3)
Evdw=Eads-Eint
(4)
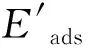
2 结果与讨论
2.1 不同类型焦组分吸附位原子的筛选
焦分子骨架结构复杂,带有较多烷基侧链,要准确计算焦组分在B酸中心附近的稳定吸附构象,首先需要找出焦组分结构中与B酸中心发生作用的原子,即吸附位原子。静电势(ESP)的定义是从无穷远处移动单位正电荷到骨架结构某原子所做的功,静电势是静电相互作用的源泉,对于理解分子间的相互作用、分子间的反应部位以及分子识别具有独到的作用,ESP为正值时,代表该处某原子对正电荷的排斥力,为负值时代表该处某原子对正电荷的吸引力,绝对值越高,说明该点的反应活性越高,可对分子的反应部位做出初步判断。笔者结合ESP电荷和焦组分在Y分子筛B酸中心附近的吸附能来确定焦组分与B酸中心产生相互作用的吸附位原子。
对碳数为11、15和19的6种结构类型焦组分碳原子进行编码,见图2,并计算各焦组分碳原子的静电势ESP,结果见表1~表3。以碳数为11的烷烃类焦组分为例,简要说明吸附位原子的筛选流程。首先根据焦组分可能的吸附构象对烷烃类焦组分的可能吸附位原子进行种类划分:骨架伯碳原子、骨架仲碳原子、骨架叔碳原子和侧链碳原子,对比各碳原子的ESP电荷,发现骨架叔碳原子的ESP电荷为正,说明它与酸中心的相互作用力为排斥力,那么该类原子作为吸附位原子的可能性低于其他类型碳原子,所以排除。然后选取具有代表性的编号为1的骨架伯碳原子、编号3的骨架仲碳原子和编号为9的侧链碳原子,并计算其在B酸中心附近的吸附能,数据表明编号为3的骨架仲碳原子作为吸附位原子时,吸附能值最大,说明此时焦组分与分子筛的B酸中心产生的相互作用较强,体系能量较低而容易稳定存在,此时焦组分的吸附构象出现概率最大。同样的计算方法来判定其他不同碳数的烷烃类、烯烃类、烷基环己烷类、烷基萘烷类、烷基苯类和烷基萘类焦组分的吸附位原子,得出以上焦组分的吸附位原子分别是仲碳原子(3、3)、双键上的仲碳原子(6、8、10)、环仲碳原子(6、6、5)、环仲碳原子(9、7、7)、芳环仲碳原子(1、1、1)、芳环仲碳原子(7、7、7)。
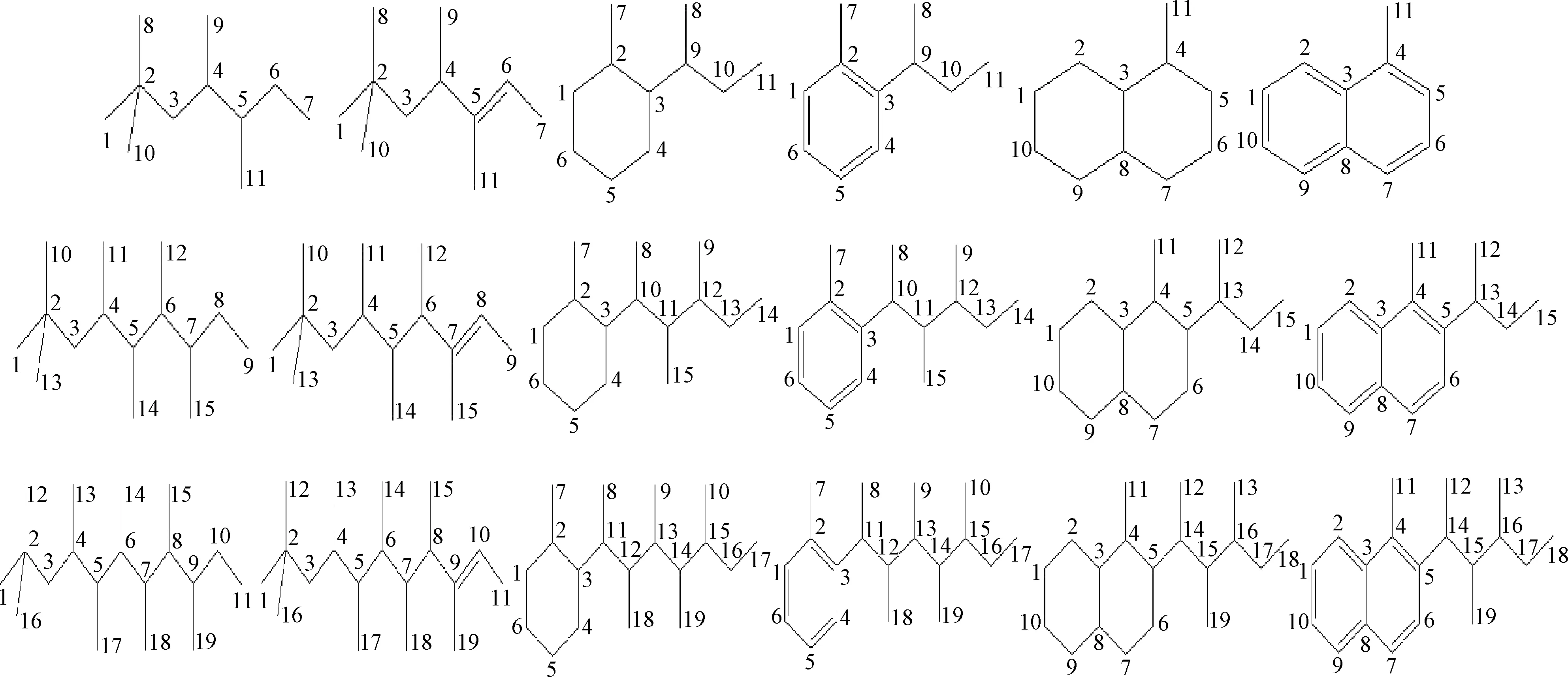
图2 焦组分模型化合物及碳原子编码示意Fig.2 Schematic diagram of coke precursor model compounds and coding of carbon atoms
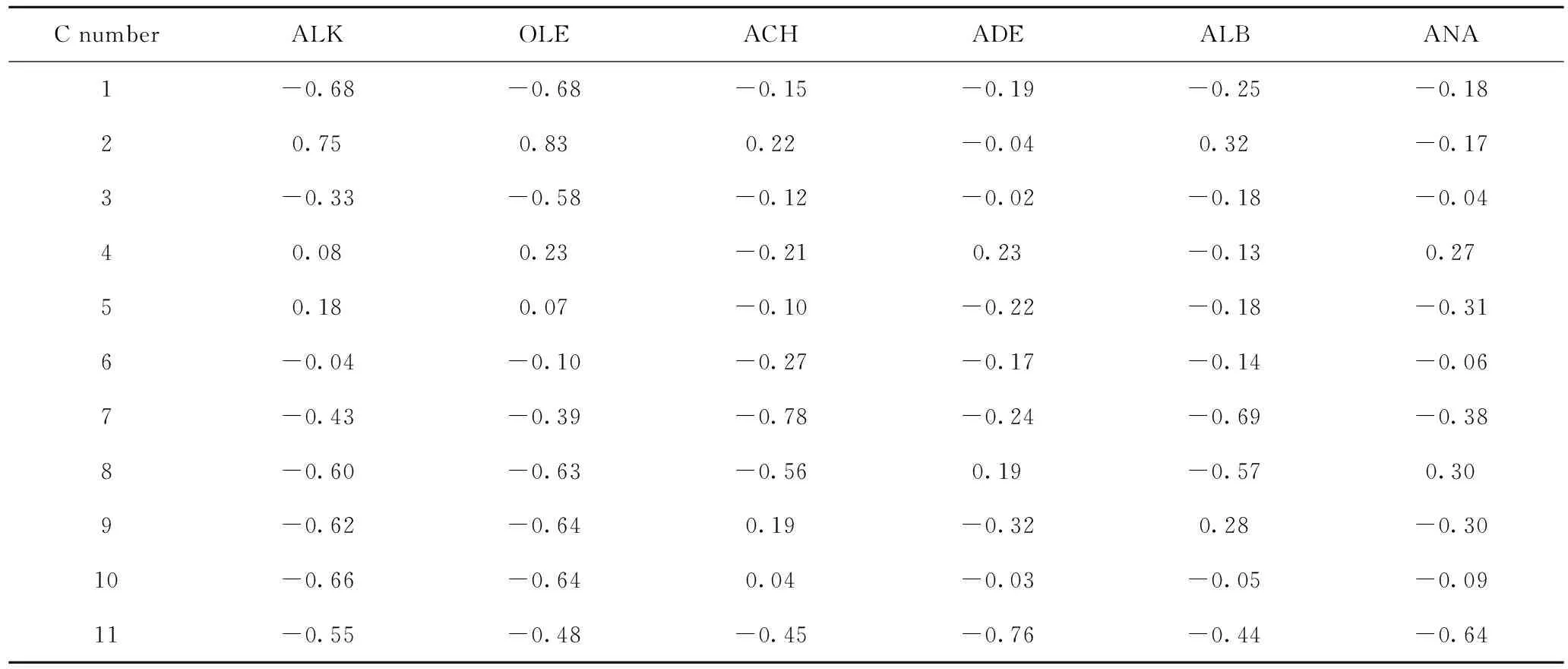
C number ALKOLEACHADEALBANA1-0.68-0.68-0.15-0.19-0.25-0.1820.750.830.22-0.040.32-0.173-0.33-0.58-0.12-0.02-0.18-0.0440.080.23-0.210.23-0.130.2750.180.07-0.10-0.22-0.18-0.316-0.04-0.10-0.27-0.17-0.14-0.067-0.43-0.39-0.78-0.24-0.69-0.388-0.60-0.63-0.560.19-0.570.309-0.62-0.640.19-0.320.28-0.3010-0.66-0.640.04-0.03-0.05-0.0911-0.55-0.48-0.45-0.76-0.44-0.64
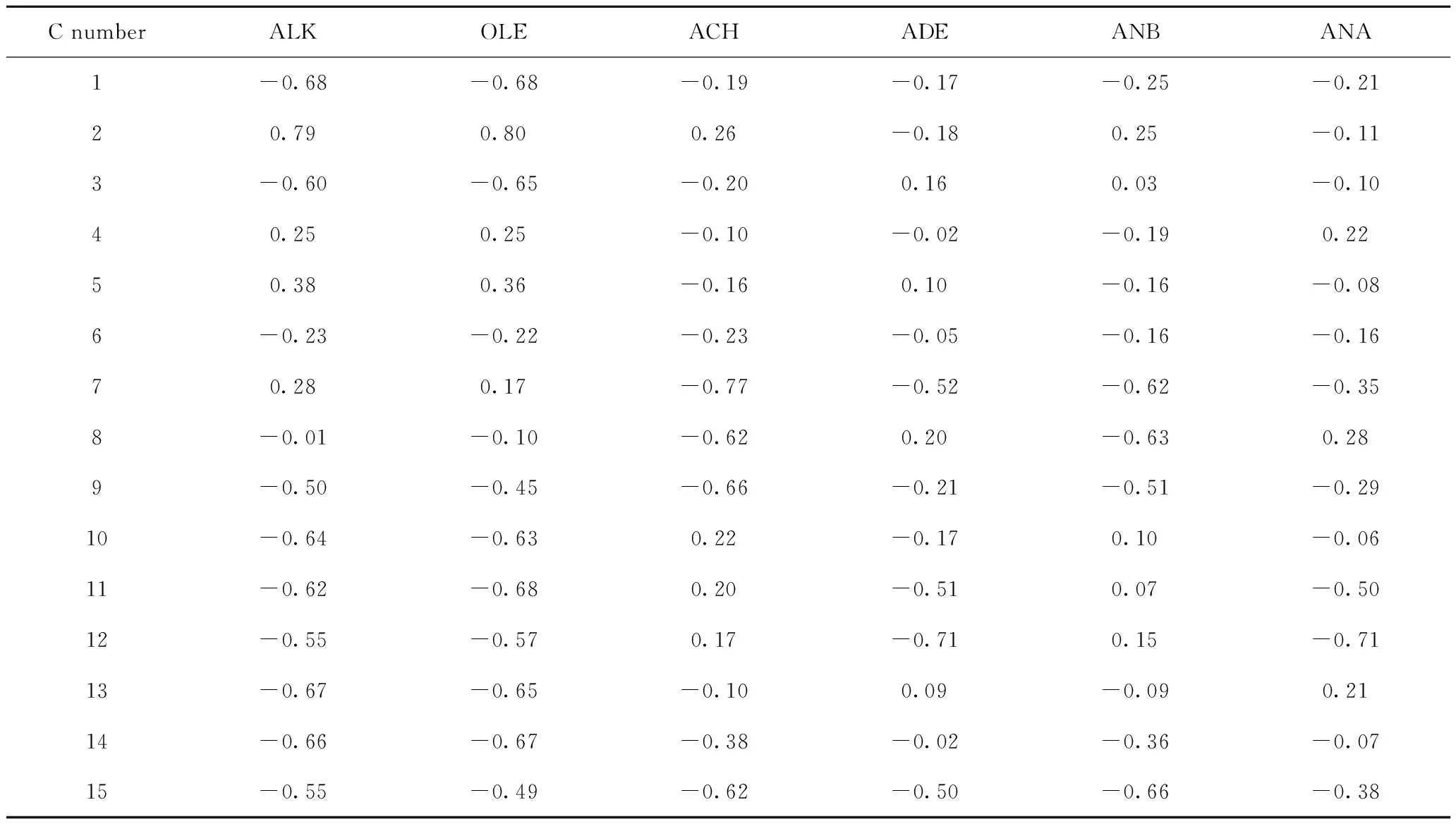
表2 碳数为15的不同结构类型焦组分的碳原子的静电势(ESP)Table 2 ESP charge of carbon atom in different structural coke precursors with carbon number of 15 ESP/V
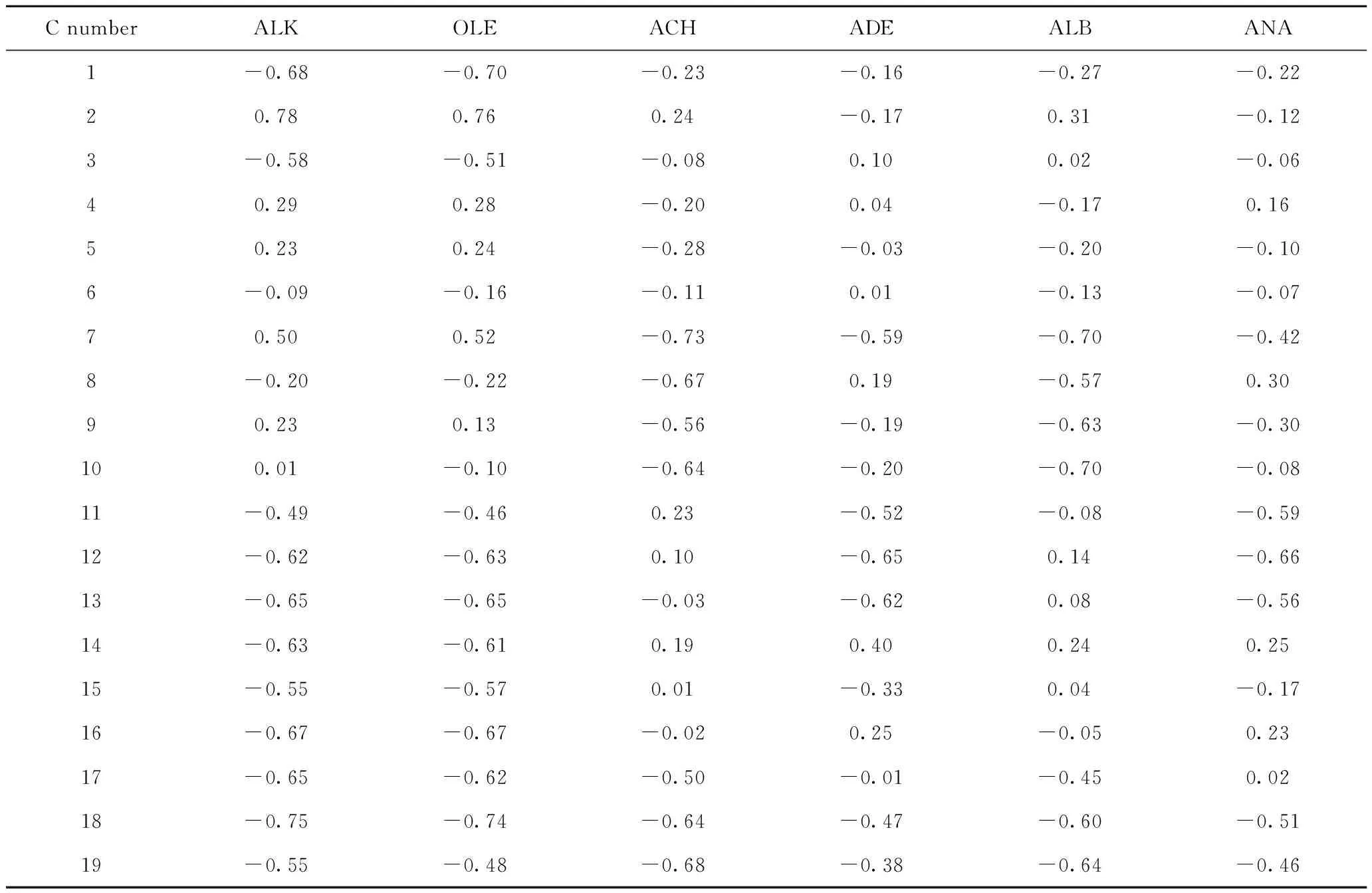
表3 碳数为19的不同结构类型焦组分的碳原子的静电势(ESP)Table 3 ESP charge of carbon atom in different structural coke precursors with carbon number of 19 ESP/V
2.2 不同类型焦组分与B酸中心的相互作用
焦组分在Y分子筛B酸中心附近吸附时,焦组分与B酸中心的氢原子发生具有电子转移的相互作用,同时焦组分与分子筛骨架原子之间也存在范德华作用,因此焦组分在分子筛孔道B酸中心附近的吸附兼具化学吸附特性和物理吸附特性。
图3为烷烃类焦组分在Y分子筛B酸中心上的吸附示意。由图3可知,烷烃类焦组分在B酸中心附近吸附时,B酸中心上的H原子通过电子诱导作用将焦组分的骨架仲碳原子及其相连的氢原子的成键电子云密度发生了极化,并得到部分电子,因此烷烃类焦组分与B酸中心相互作用的形式是电子诱导作用。
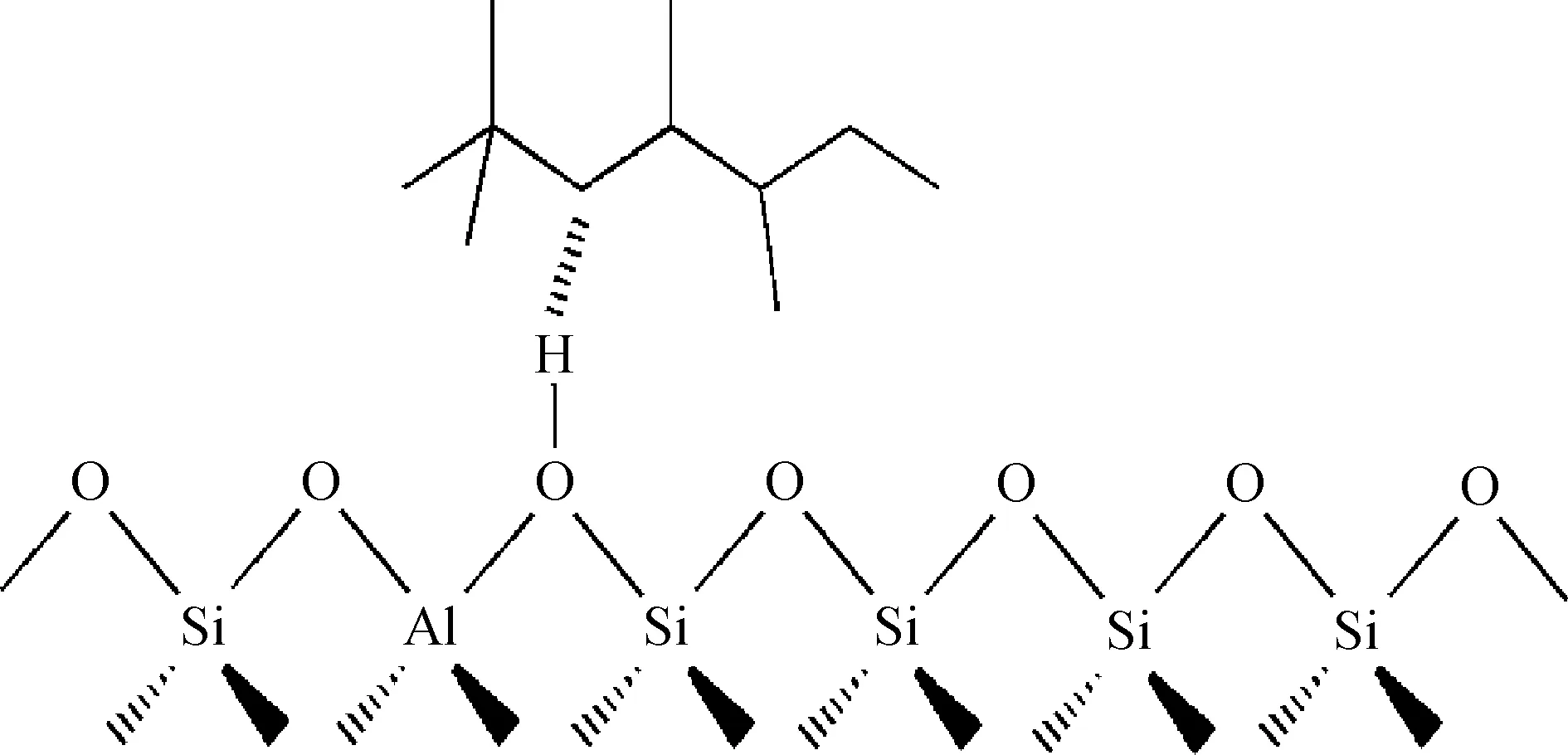
图3 烷烃类焦组分在Y分子筛B酸中心上的吸附示意Fig.3 Adsorption diagram of alkanecoke precursors on B acid site in Y zeolite
图4为烯烃类焦组分在Y分子筛B酸中心上的吸附示意。由图4可知,烯烃类焦组分在B酸中心吸附时,烯烃双键上的π电子的P轨道与B酸中心的氢原子缺电子轨道发生重叠,导致B酸中心的氢原子与双键上的碳原子共享双键上的π电子,因此烯烃类焦组分与分子筛B酸中心的相互作用形式是π-H共价键。
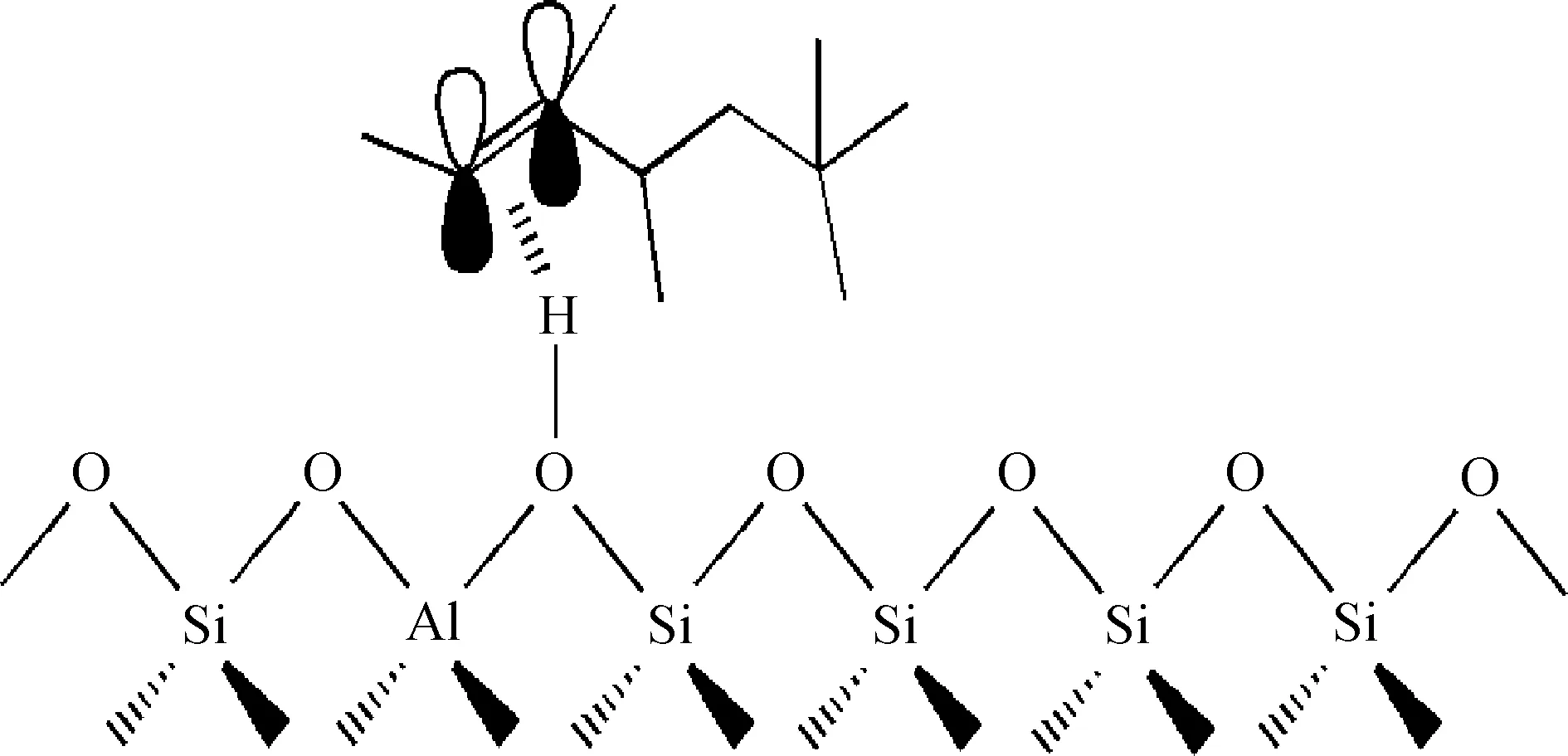
图4 烯烃类焦组分在Y分子筛B酸中心上的吸附示意Fig.4 Adsorption diagram of olefin coke procures on B acid site in Y zeolite
图5为环烷类焦组分在Y分子筛B酸中心上的吸附示意。由图5可知,烷基环己烷类和烷基萘烷类焦组分在B酸中心附近吸附时,与烷烃类焦组分的吸附过程类似,其与分子筛B酸中心相互作用形式同为电子诱导作用,酸中心上的H原子通过电子诱导作用将焦组分的环仲碳原子及其相连的氢原子的成键电子云密度发生了极化,并得到部分电子。
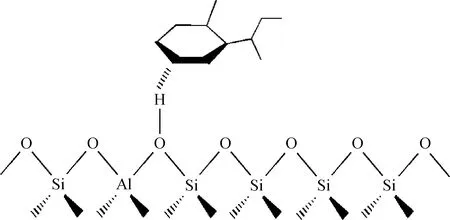
图5 环烷类焦组分在Y分子筛B酸中心上的吸附示意Fig.5 Adsorption diagram of naphthenic coke precursors on B acid site in Y zeolite
图6为芳香类焦组分在Y分子筛B酸中心上的吸附示意。由图6看到,烷基苯类和烷基萘类焦组分在B酸中心附近吸附时,苯环上的π电子的P轨道与B酸中心上的氢原子缺电子轨道重叠,导致B酸中心的氢原子与环仲碳原子共享苯环上的π电子,因此芳香类焦组分与B酸中心的相互作用形式为π-H共价键。
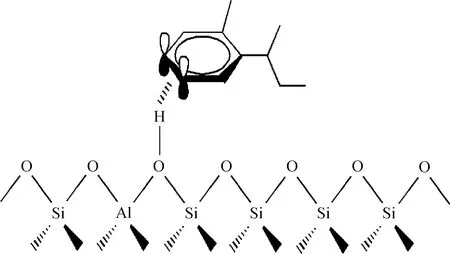
图6 芳香类焦组分在Y分子筛B酸中心上的吸附示意Fig.6 Adsorption diagram of aromatic coke precursors on B acid site in Y zeolite
2.3 不同烃类焦组分的吸附构象
2.3.1 直链类
以碳数为11、15和19的烷烃类和烯烃类焦组分为模型化合物,计算直链类焦组分在Y分子筛孔道内B酸中心附近的稳定吸附构象,为便于对比分析吸附构象对分子筛孔道内空间的占据情况,将直链类焦组分的主链骨架碳原子进行适度放大并忽略侧链原子对图示效果的影响,并分别给出X、Y、Z3个视角方向的图像,如图7所示。从图7可以明显看出烷烃类和烯烃类焦组分在分子筛孔道内B酸中心附近的稳定吸附构象类似,在Y方向上,随着碳数的增加,烷烃类和烯烃类焦组分的主链骨架碳原子对附近轴向相邻的两个十二元环孔道空间均无占据,但在X和Z方向上,2种焦组分对所在的十二元环孔道空间具有明显的占据,尤其是在X方向视角下,随着碳数的增加,焦组分对所在的十二元环孔道的径向空间占据也越大。以上现象表明,烷烃类和烯烃类焦组分在Y分子筛孔道内B酸中心附近吸附后,对分子筛的三维孔道呈二维空间占据,且随着碳数的增加,在X方向上对所在十二元环孔道的径向空间占据也越大。
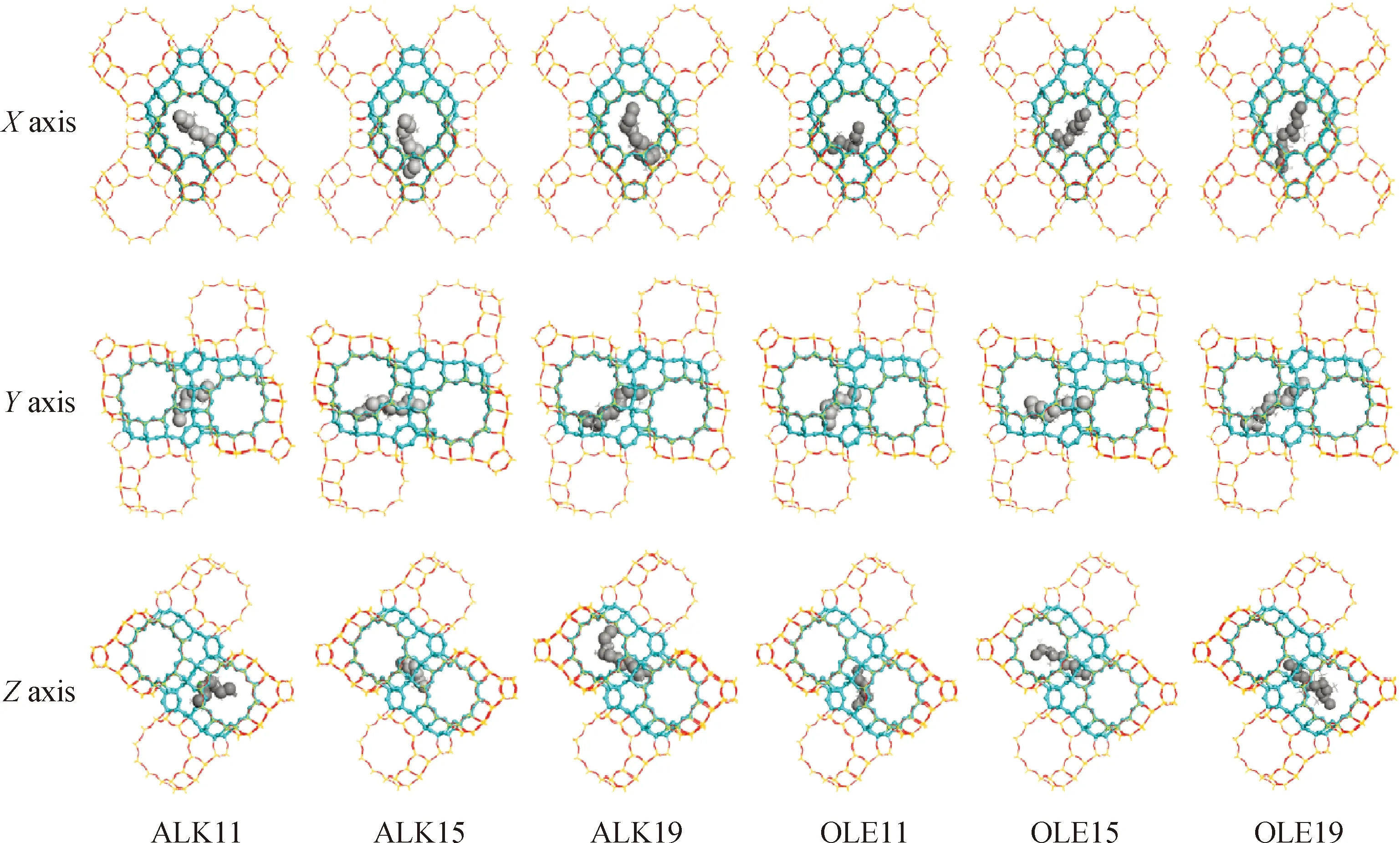
图7 直链类焦组分在Y分子筛孔道内的稳定吸附构象示意Fig.7 Adsorptive conformation of straight chain coke precursors in Y zeolite channel
2.3.2 环状类
以碳数为11、15和19的烷基环己烷、烷基萘烷、烷基苯和烷基萘类焦组分为模型化合物,计算环状类类焦组分在Y分子筛孔道内B酸中心附近的稳定吸附构象,如图8和图9所示。从X、Y、Z3个视角来看,环状类焦组分吸附后对分子筛的三维十二元环孔道空间均有占据,且随着碳数的增加,占据的空间也越大。在X方向上,焦组分的环状结构与所在的十二元环孔道的轴向呈一定夹角,随着碳数的增加,夹角有增大的趋势。对比单环结构和双环结构类焦组分的稳定吸附构象,可以发现双环类焦组分与所在的十二元环孔道的轴向夹角更大,随着碳数的增加,夹角的增大趋势也更明显,而烷基萘类焦组分的轴向夹角一直接近90°,这说明烷基萘类焦组分在X方向上对所在十二元环孔道的径向空间占据也更大。另外随着碳数的增加,单环类焦组分的侧链长度增加,并向相邻的超笼空间延伸。以上现象表明,环状类焦组分在Y分子筛孔道内B酸中心附近吸附后,对分子筛的三维孔道呈三维空间占据,且在X方向上,随着碳数的增加,相比单环类焦组分,双环类焦组分对所在十二元环孔道的径向空间占据更为明显,堵孔效应也更大,其中烷基萘类焦组分的堵孔效应最大。
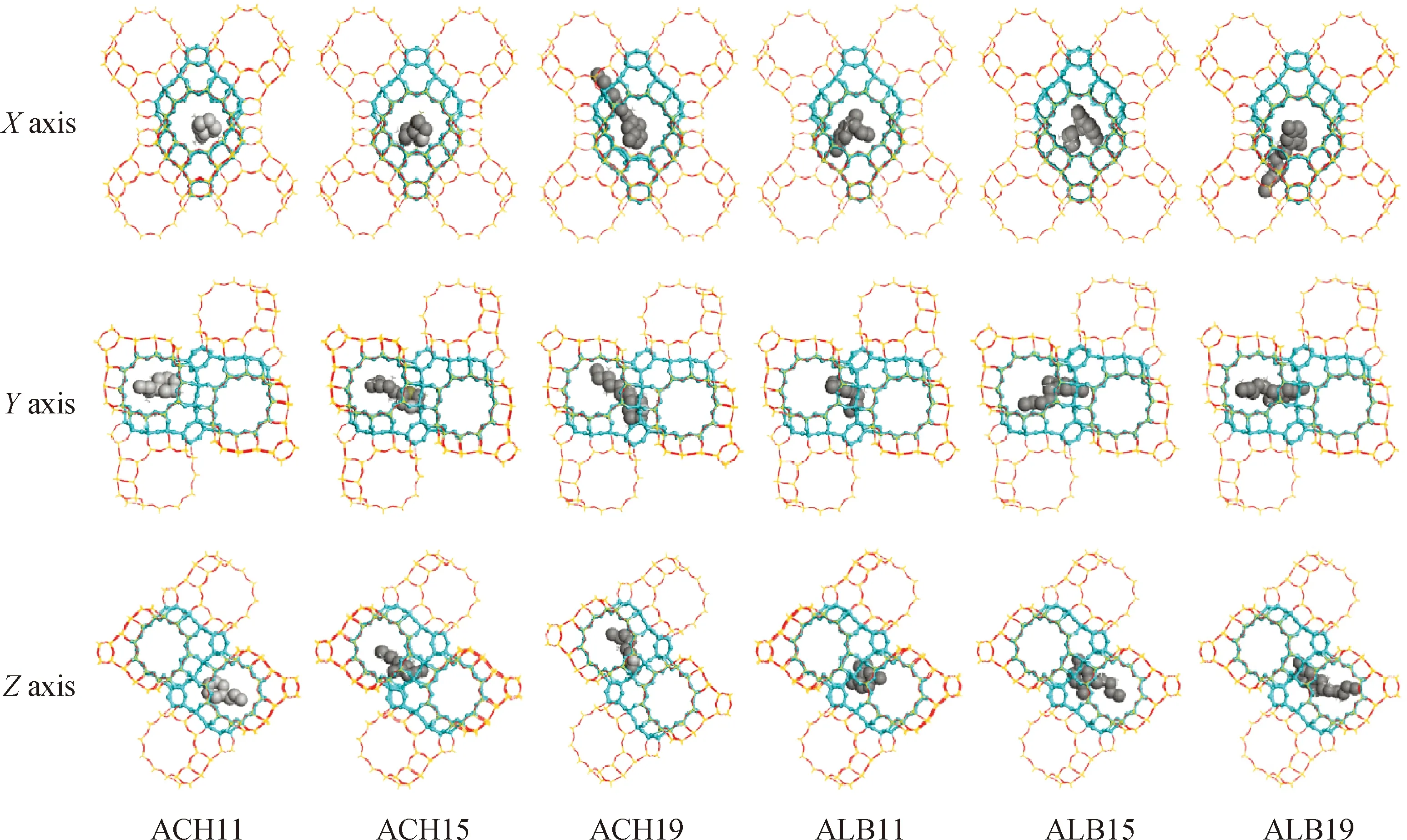
图8 单环类焦组分在Y分子筛孔道内的稳定吸附构象示意Fig.8 Adsorptive conformation of monocyclic coke precursors in Y zeolite channel
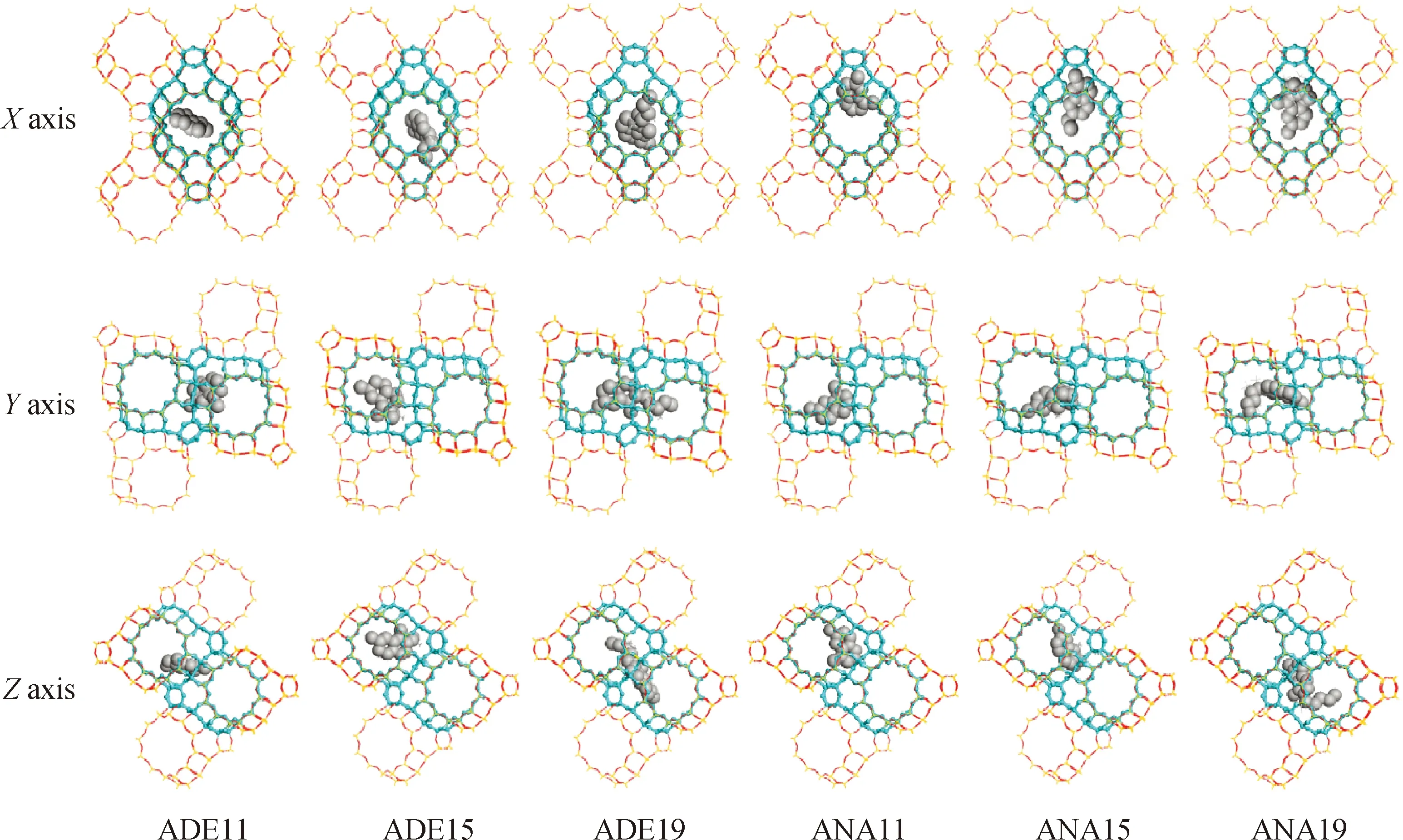
图9 双环类焦组分在Y分子筛孔道内的稳定吸附构象示意Fig.9 Adsorptive conformation of dicyclo coke precursors in Y zeolite channel
2.4 不同结构类型焦组分的吸附性能比较
综合不同结构类型焦组分在分子筛孔道内吸附数据,计算其吸附能值、范德华作用能和与Y分子筛B酸中心的相互作用能,如图10所示。从图10可以明显看出,不同结构类型焦组分在分子筛孔道内的吸附能和范德华作用能均随着碳数的增加而增大,且两者的增幅相近,而焦组分与B酸中心的相互作用能则基本保持不变,由此可看出焦组分碳数的增加对焦组分与B酸中心之间的相互作用不产生影响,而与分子筛骨架原子间产生范德华作用的焦组分的原子总数的增加,是焦组分在分子筛孔道内吸附能值升高的内在诱因。另外,不同结构类型焦组分与B酸中心之间的相互作用存在差异性,其由大到小的顺序为烯烃类、烷基萘类、烷基苯类、烷烃类、烷基环己烷类、烷基萘烷类,而焦组分的吸附能由大到小的顺序为烯烃类、烷基苯类、烷基萘类、烷烃类、烷基环己烷类、烷基萘烷类。
3 结 论
(1)分子筛催化异丁烷/丁烯烷基化反应体系中,同碳数不同结构类型焦组分在Y分子筛孔道内B酸中心附近吸附时,吸附由强到弱的顺序为烯烃类、烷基苯类、烷基萘类、烷烃类、烷基环己烷类、烷基萘烷类。
(2)随着碳数的增加,不同结构类型焦组分在Y分子筛孔道内的吸附能和范德华作用能逐渐增大,增幅相近,而与B酸中心的相互作用能基本保持不变。不同类型的焦组分与B酸中心的相互作用存在差异性,由强到弱的顺序为烯烃类、烷基萘类、烷基苯类、烷烃类、烷基环己烷类、烷基萘烷类。
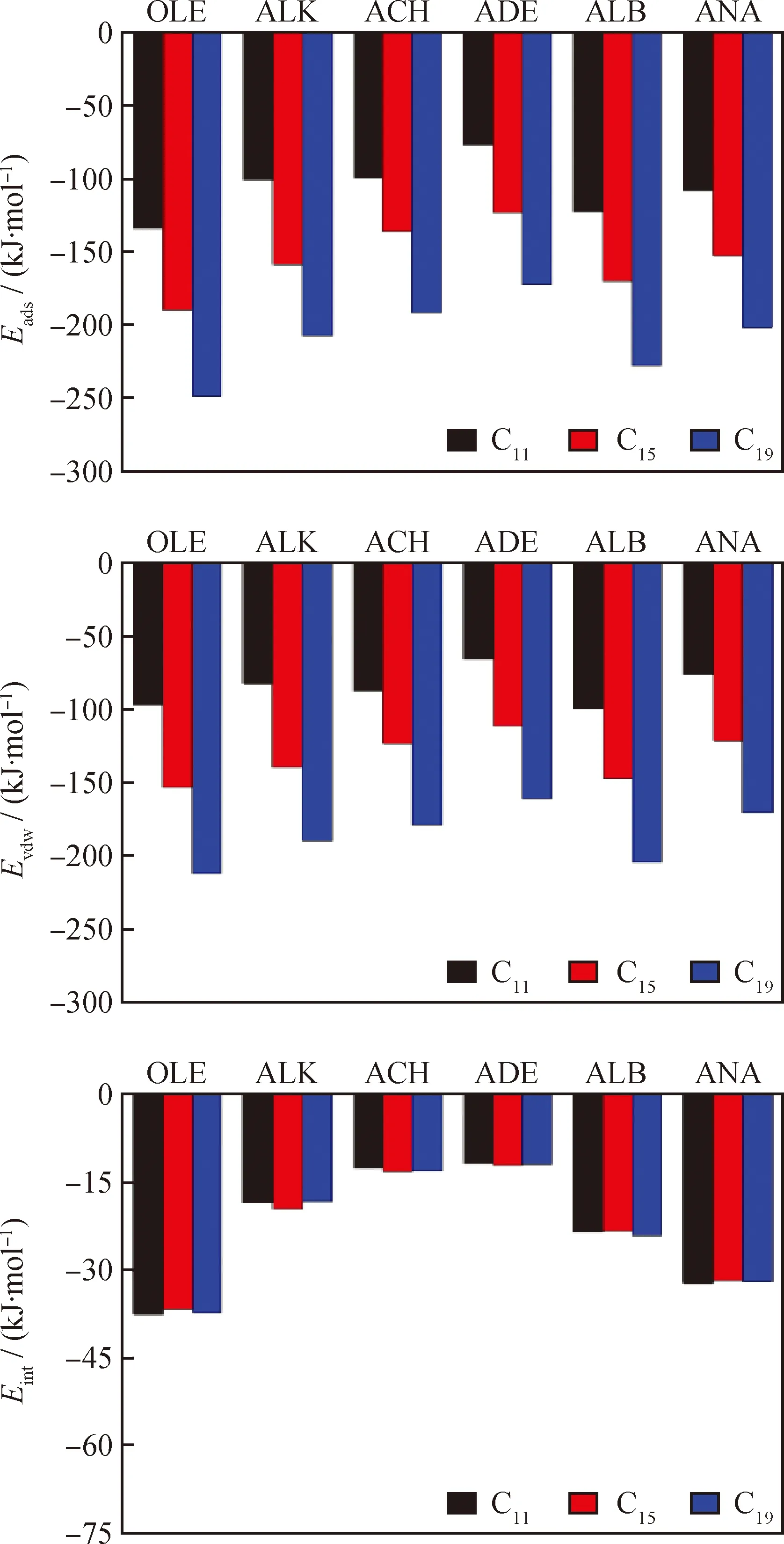
图10 不同碳数及结构类型的焦组分的吸附能、范德华作用能、与B酸中心的相互作用能值变化Fig.10 Changes of adsorption energy, VDW and interaction energy with B acid cite for coke precursors with different carbon numbers and structures
(3)烷烃类和烯烃类焦组分对Y分子筛的三维十二元环孔道呈二维空间占据,烷基环己烷类、烷基苯类、烷基萘烷类和烷基萘类焦组分对Y分子筛的十二元环孔道呈三维空间占据。在X方向上,相比单环类焦组分,双环类焦组分对分子筛十二元环孔道的径向空间占据更明显,堵孔效应更大,其中烷基萘类焦组分的堵孔效应最为突出。
