胚鼠腭裂形成过程中腭胚组织全基因组DNA甲基化的研究*
2018-09-27舒申友董泽君
刘 丹, 舒申友, 李 珂, 舒 璇, 董泽君, 郎 兴
(汕头大学医学院第二附属医院烧伤整形外科, 广东 汕头 515041)
非综合征型单纯性腭裂(nonsyndromic cleft pa-late only,NSCPO)是一种先天性缺陷病,严重影响人们的健康及生活质量。目前NSCPO的发病机制尚不清晰,已证实基因突变和环境影响都是NSCPO的复杂病因[1-2]。大量研究表明DNA甲基化对维持细胞正常功能、胚胎发育和肿瘤的发生有重要作用,因作用位点较广且调控结果可逆、可遗传等特性而受到关注。Rogers等[3]的研究表明,对妊娠第10天(gestational day 10, GD10)的雌性孕鼠给予高剂量的DNA去甲基化剂5-氮杂-2’-脱氧胞苷时,胚鼠腭裂的频率变高。Bulut等[4]注意到,在GD11和GD14向小鼠施用5-氮杂胞苷(5-氮杂-2’-脱氧胞苷的核苷类似物)时出现多种先天性发育畸形,其中就包括腭裂,提示该化合物的去甲基化作用对早期胚胎发育具有易感性。因此本研究使用高效低成本全基因组DNA甲基化检测技术——甲基化修饰依赖性内切酶测序(methylation-dependent restriction-site associated DNA sequencing,MethylRAD-Seq)法对不同时段的腭裂模型组和正常对照组小鼠腭胚组织进行全基因组DNA甲基化测序,比较基因组不同元件中甲基化位点的差异分布及甲基化水平的差异基因,对甲基化位点的位置信息进行注释,并对差异甲基化位点相关基因和甲基化差异基因进行GO功能富集和KEGG通路富集分析,筛选与胚鼠腭裂形成相关基因及信号通路,为进一步阐明NSCPO的发病机制和进一步实验,提供理论依据。
材 料 和 方 法
1 动物
SPF级C57BL/J小鼠动物许可证编号为SCXK(京)2012-0001(北京维通利华公司),饲养于汕头大学医学院SPF级实验动物房,8~10 周龄,体重22~28 g,饲养温度(25±2) ℃,人工调控昼夜循环,自由进饮进食。
2 主要试剂
全反式维甲酸(all-transretinoic acid,ATRA)(Sigma);玉米油(中粮集团)。
3 主要方法
3.1构建维甲酸诱导的胚鼠腭裂模型 将70 mg ATRA溶于10 mL 玉米油中,配制浓度为7 g/L的ATRA溶液,-20 ℃保存备用。实验当天中午12:00以2∶1雌雄合笼,次日晨8:00将雌雄鼠分笼饲养,雌鼠均视为疑似受孕,记为GD0.5。雌鼠随机编号,称体重并记录。在GD10.5 晨 8:00 时依次称量雌鼠体重,观察腹部明显膨隆且体重增加大于2.5 g者视为实验孕鼠,其余视为假孕并继续以同法合笼交配,共得到的实验孕鼠18只,随机分3组(A、 B和C组),每组6只;每组再分成2小组,每小组3只,比如A组包括A1、A2、A3、a1、a2和a3,其中A为模型组,a为对照组。所有模型组(A1、A2、A3、B1、B2、B3、C1、C2和C3)在GD10.5分别给予管饲ATRA (70 mg/kg),对照组(a1、a2、a3、b1、b2、b3、c1、c2和c3)在GD10.5给予管饲玉米油(0.20 mL/kg)。
3.2样本的获取及保存 分别于GD13.5、14.5和16.5使用脱颈法处死模型组及对照组小鼠,无菌操作,在显微镜下解剖摘取胚鼠口腔内的腭突组织,新鲜腭突组织标本取下立即放入无菌冻存管并置于液氮冻存(来自同1个孕鼠的所有腭突组织作为1例样本)。
3.3全基因组DNA甲基化测序 采用QIAamp DNA Mini Kit 法提取组织样本DNA,并采用 1%的琼脂糖凝胶电泳检测样本DNA纯度,18个样本委托上海欧易生物医学科技有限公司进行全基因组DNA甲基化检测。MethylRAD-Seq法是利用甲基化修饰依赖性内切酶(FspE I)对基因组DNA进行酶切,FspE I可以识别DNA上甲基化位点,把FspE I的识别位点标识为CCGG和CCWGG,其中简并碱基W代表碱基A和T,在识别位置的下游隔一定位置进行酶切,如果DNA 2条链的甲基化状态是对称的,那么就可以切割产生一个固定长度的片段(32 bp),这些片段给予合适的接头序列(adaptor)连接后进行PCR扩增构建成带有样本特异性条纹码序列(barcode sequence)的文库。18个样品的标签序列文库在Hiseq X Ten平台进行高通量测序,得到原始测序序列(Raw Data)。原始测序序列经过滤删除含有接头序列的Reads及低质量Reads,使用Pear 0.9.6软件拼接后,最后过滤删除含有N碱基的序列得到Clean Data。参考基因组序列数据库(ftp://ftp.ensembl.org/pub/release-84/fasta/mus_musculus/dna/Mus_musculus.GRCm38.dna.toplevel.fa.gz),从C57BL/J小鼠基因组序列中提取所有的CCGG和CCWGG位点,建立标签对比的参考序列。最后利用SOAP 2.21软件将这些高质量的MethylRAD测序标签比对到构建的CmCGG和CmCWGG位点的参考序列上(参数设置为:-M4-v 2-r0)唯一位置的序列,进行全基因组DNA甲基化水平的检测,将测序深度不低于3的位点判定为可靠的甲基化位点。最后将测得的3个时段有差异甲基化位点所在的基因及甲基化差异基因都导入DAVID基因功能分析平台进行GO和 KEGG通路分析,对基因的功能及信号通路进行注释。
结 果
1 甲基化位点的数量及甲基化水平的比较
根据等长标签扩增效率的一致性,甲基化标签的测序深度可以反映位点的甲基化水平。以测序深度不低于3的位点判定为可靠的甲基化位点,考虑到参考序列中存在重复区,位于重复区的Reads能够同时比对到参考序列的多个位置,对于这种甲基化位点的实际发生位置难以进行判断,鉴于此,在进行比对时,不考虑重复比对的Reads,18个文库的Enzyme Reads (Clean Reads 质控后含有预期酶切位点的Reads)能比对到参考序列唯一位置的比对率见表1。根据这18个样本的全基因组DNA甲基化的数据,统计分析了每个样本的甲基化位点数量及平均测序深度,见表2。
表118个文库的EnzymeReads比对到参考序列唯一位置的比对率
Table 1. The ratio of Enzyme Reads matching the alignment to the only position in the reference sequence

SampleClean ReadsEnzyme ReadsMapping ReadsRatioA1142,855,38380,720,50857,927,32571.76%A2142,855,38379,838,18659,697,33474.77%A3142,855,38378,129,09256,390,99572.18%B1131,029,92052,959,69138,391,98672.49%B2131,029,92056,224,18141,472,60573.76%B3131,029,92054,365,63438,595,47370.99%C1154,938,89169,209,73550,154,76972.47%C2116,896,71535,423,26724,085,45567.99%C3116,896,71544,807,99532,803,91173.21%a1116,896,71546,276,55434,624,76874.82%a2116,896,71542,914,10430,874,53171.94%a3142,855,38371,659,10652,037,53372.62%b1142,120,40259,873,38744,498,70374.32%b2142,120,40258,784,35842,436,66072.19%b3131,029,92042,987,51629,595,82668.85%c1154,938,89155,011,78437,939,28368.97%c2154,938,89168,822,66149,747,69972.28%c3154,938,89171,858,94053,483,39974.43%
表218个样本甲基化位点平均测序深度
Table 2. The average sequencing depth of the methylation sites (CCGG and CCWGG) in 18 samples

SampleCCGG siteCCWGG siteSite numberMean depthSite numberMean depthA1812,06953.0991,6529.94A2869,98048.55105,9989.22A3837,89049.05108,1589.29B1752,15038.6057,5459.07B2815,02237.1250,8988.33B3770,02637.6555,4698.32C1818,90944.9369,6508.85C2742,80924.7449,2277.99C3736,46332.9953,1678.17a1784,09130.0750,5777.63a2761,07230.0856,4357.87a3850,65746.9394,7848.56b1823,50838.2978,5788.15b2790,14240.3269,1228.65b3767,88829.1657,2528.36c1828,04334.9165,3609.07c2800,21747.1763,6529.68c3860,56345.3474,8778.33
2 甲基化位点在基因元件的分布情况
使用SnpEff 4.3p软件,根据注释文件计算获得非翻译区(untranslated region, UTR),使用bedtools 2.25.0软件统计了18个样本的甲基化位点在3’-UTR、5’-UTR、TSS2000(转录起始位点上游2 000 bp内的区域)、intron(内含子区)、exon(外显子区)和intergenic region (基因间区)6个基因功能元件的甲基化位点数、分布特点及差异,甲基化位点数量在exon、intron和intergenic region这3个基因元件上的数量明显多于3’-UTR、5’-UTR和TSS2000这3个元件,差异有统计学意义(P<0.05),见图1。

Figure 1. The distribution and quantitative statistics of 18 samples of DNA methylation sites in 6 gene functional elements. A: CCGG methylation sites; B: CCWGG methylation sites.
图118个样本DNA甲基化位点在6个基因功能元件的分布及数量统计
3 差异甲基化位点在基因及染色体上的分布特点
以实验时点分3组进行比较,使用edgeR软件计算各个位点在组间的差异P值及差异倍数log2FC并判断甲基化水平变化趋势,进行统计检验后,将P<0.05且log2FC绝对值大于1的位点判定为差异甲基化位点。3个时点组的差异甲基化位点数量见表3,在不同基因功能元件上所占的比例见表4。从上述2个表中可见3个时点的差异甲基化位点都主要集中在内含子区、基因间区和外显子区,组间比较无显著差异。有差异的甲基化位点主要集中在CCGG位点上;从时间上比较,随着时间推移,上调的差异甲基化位点数量明显减少,下调的差异位点数量逐渐增加。差异甲基化位点在染色体上的分布可能是不均匀的,在基因组不同功能元件的差异甲基化位点分布表明,CCGG/CCWGG主要位于内含子和基因间区域,见图2。
表33个时点差异甲基化位点的数量统计
Table 3. The numbers of differential methylation sites at the 3 time points
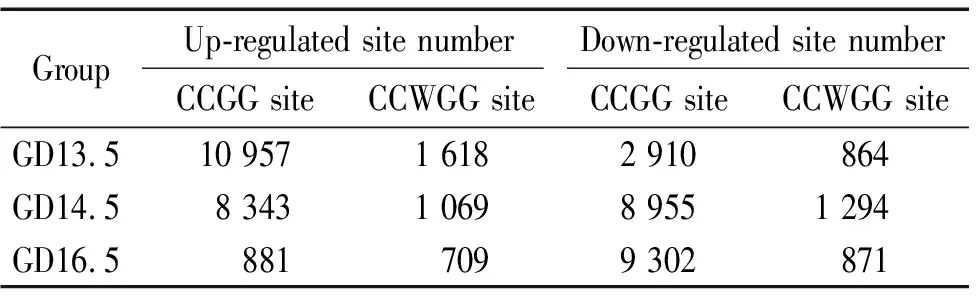
GroupUp-regulated site numberDown-regulated site numberCCGG siteCCWGG siteCCGG siteCCWGG siteGD13.510 9571 6182 910864GD14.58 3431 0698 9551 294GD16.58817099 302871

表4 3个时点差异甲基化位点在基因功能元件上所占的比例

Figure 2. The distribution of differential methylation sites on chromosomes. A, C and E: CCGG methylation sites; B, D and F: CCWGG methylation sites. A and B: GD13.5; C and D: GD14.5; E and F: GD16.5. The methylation level up-regulation and down-regulation sites are expressed in brown and blue, respectively.
图2差异甲基化位点在染色体上的分布图
4 差异甲基化位点相关基因的GO和KEGG富集分析
对差异甲基化位点所在的基因进行GO功能富集分析,对基因的功能进行描述(结合GO注释结果),GO分析的结果结合生物学意义从而挑选用于后续研究的基因。利用KEGG数据库对差异位点所在基因进行通路分析(结合KEGG注释结果),通路分析对实验结果有提示的作用,通过差异位点的基因进行通路分析,可以找到差异位点的基因可能与哪些细胞通路的改变有关。我们统计并绘制了差异甲基化位点相关基因的GO功能富集前30项及KEGG通路前20项的富集图,见图3、4。
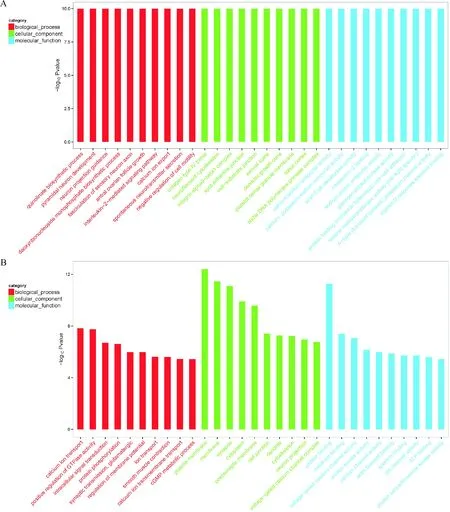
Figure 3. Differential methylation site-related genes analyzed by GO function enrichment. A: CCGG methylation sites; B: CCWGG methylation sites.
图3差异甲基化位点相关基因的GO功能富集结果
5 甲基化差异基因的GO和KEGG富集分析
计算基因内的所有位点测序深度总和,代表其基因的甲基化水平,而后进行组间比较,分组同前。对模型组和对照组样品进行统计检验的基因,要求至少存在一个组,组内所有样品在基因的测序深度均高于3,对P<0.05且log2FC绝对值大于1的位点判定为差异基因。我们汇总了这些甲基化水平有差异的基因,统计了这些基因甲基化水平上、下调的信息,绘制了差异甲基化基因的数量统计表,见表5。最后将这些甲基化差异基因进行GO功能富集和 KEGG信号通路富集分析,为寻找腭裂相关基因及通路提供数据依据。我们统计并绘制了甲基化差异基因GO功能富集的前30项及KEGG通路前20项的富集图,见图5、6。
6 腭裂形成相关的基因及信号通路
目前普遍认为ATRA导致小鼠腭裂的发生机制主要有以下3点:(1)内侧边缘上皮细胞的周皮细胞发生凋亡所致;(2)原舌肌及下颌骨在发育过程中相应细胞发生凋亡所致;(3)抑制间充质细胞的增殖所致。参照上述病因机制,本实验选取了数个明显GO富集结果和KEGG通路分析结果,并在其中挑取了6个有明显差异的基因(Fgf16、Jarid2、Kdm6a、Nr3c1、Tbx22和Trp53),以及MAPK、Wnt和TGF-β信号通路作为下一步验证对象,见表6。

Figure 4. Differential methylation site-related genes analyzed by KEGG pathway enrichment. A: CCGG methylation sites; B: CCWGG methylation sites.
图4差异甲基化位点相关基因的KEGG通路富集结果

表5 3个时点差异甲基化基因的数量统计

Figure 5. Methylation differential genes analyzed by GO function enrichment. A: CCGG methylation sites; B: CCWGG methylation sites.
图5甲基化差异基因的GO富集结果
讨 论
表观遗传学与腭裂形成的相关性的研究最近几年才开始引起关注,表观遗传学包括DNA甲基化、microRNA、组蛋白修饰、染色质重塑及非编码 RNA调控等。目前研究腭裂发生机制主要集中在基因突变和环境变化。遗传改变被认为可导致20%~25%的腭裂病例[5-6],而解释遗传改变的一种机制是在胚胎发育过程中易感基因的异常甲基化,导致正常腭发育所必需的关键基因表达发生改变,故查找腭裂疾病相关基因及探究其致病机制尤为迫切。
本实验我们选用ATRA诱导的小鼠腭裂模型和MethylRAD-Seq技术。在腭裂疾病的基因研究中,常用到ATRA、地塞米松和TCDD(四氯二苯并-p-二噁英)等药物诱导先天性小鼠腭裂模型。本实验我们选用ATRA诱导C57BL小鼠,大量动物实验证明此模型稳定可靠[7]。MethylRAD-Seq技术是一种新开发的高效低成本的全基因组的DNA甲基化检测技术[8],可以对全基因组内的甲基化位点进行定量和定性分析,其原理主要是利用甲基化修饰依赖性内切酶(FspE I和MspJ I)对基因组DNA进行酶切,基因组中CmCGG和CmCWGG位点上的甲基化修饰均可以被识别,酶切后产生具有核心甲基化位点的等长标签,对标签文库再进行高通量测序,最终获得全基因组范围内的甲基化位点序列信息。根据等长标签扩增效率的一致性,甲基化标签的测序深度可以反映位点(CmCGG/CmCWGG)的甲基化水平。已有研究利用基因组背景清晰的模式生物拟南芥,对MethylRAD-Seq技术进行方法学验证[9]。结果表明,MethylRAD-Seq具有很好的技术重复性,假阳性率在0.5%以内,可以检测全基因组范围内胞嘧啶的甲基化水平,并且该方法可以评估基因组染色体区域的甲基化水平分布。该技术结合了甲基修饰依赖性内切酶和简化基因组限制性酶切分型技术的特点,在不需要基因组背景信息的前提下可以大规模挖掘全基因组范围内的甲基化位点,直接获得甲基化胞嘧啶的序列信息;既可以用于未知甲基化位点的开发,也可用于不同样本间的甲基化位点的差异研究。

Figure 6. Methylation differential gene by KEGG pathway enrichment results. A: CCGG methylation sites; B: CCWGG methylation sites.
图6甲基化差异基因的KEGG富集结果

表6 Fgf16、Jarid2、Kdm6a、Nr3c1、Tbx22和Trp53基因注释信息
FC: fold change; Chr: chromosome.
本实验统计发现了样本的DNA差异甲基化位点主要分布在内含子区、基因间区和外显子区。内含子区域的差异甲基化位点占到了约39%,这可能是由于内含子对翻译转录产物无结构意义,不受自然选择的压力,累积更多突变有关。与甲基化使基因表达降低的启动子区相反,基因间区的甲基化水平倾向于与转录正相关[10],基因间区的甲基化与转录的激活机制密切相关[11]。本次测序数据提示基因间区的差异甲基化位点占到了约40%。但DNA甲基化在基因间区内的具体作用尚不清楚,组织特异性基因的基因间区中发生异常甲基化,是否可能减少转录或增加转录延伸;基因间区的甲基化修饰在调节转录功能上是否可部分替代启动子的作用值得我们进一步探究。通过对3组18个样本测序数据进行分析,我们发现基因不同功能元件的甲基化位点的峰值个数和峰值在对照组无明显变化,实验组甲基化整体水平随着时间推移逐渐降低;实验组和对照组的CmCGG型甲基化位点都明显多于CmCWGG型,而且CmCGG型的甲基化位点的甲基化测序深度明显高于CmCWGG型,推断基因组内DNA发生甲基化的位点,仍主要集中在CpG-二核苷酸上。甲基化差异基因的分析证实了intron、exon和intergenic region这3个功能元件上甲基化差异基因数量明显多于3’-UTR、5’-UTR和TSS2000,我们推测相关基因的异常甲基化可能是腭裂发生的重要原因。
关于腭裂与DNA甲基化的研究,相关甲基化差异基因筛选和验证以及相关通路的调控机制将是非常重要的步骤。本实验通过对3组18个样本进行测序,发现3个组共计1 497个有差异甲基化表达的基因(P<0.05),并进行GO功能富集和 KEGG通路富集分析,结合胚鼠腭裂病理生理发生机制,筛选了腭裂形成相关的6个基因(Fgf16、Jarid2、Kdm6a、Nr3c1、Tbx22和Trp53);这些基因的差异甲基化位点主要位于exon、TSS2000、intron和intergenic region;结合这些基因对应的GO注释,本实验发现Fgf16与GO:0005104(成纤维细胞生长因子受体结合的分子功能)和GO:0008543(成纤维细胞生长因子受体信号通路)密切相关;Jarid2与GO:0031061(组蛋白甲基化的负调控)、GO:0048863(干细胞分化)、GO:0042127(细胞增殖的调控)和GO:0016568(染色质修饰)密切相关;Tbx22与GO:0005634(细胞核)、GO:0045892(以DNA为模板转录的负调控)、GO:0000122(RNA聚合酶II对转录的负调控)、GO:0003700(DNA结合转录因子活性)、GO:0006351(以DNA为模板的转录)和GO:0003677(DNA结合的分子功能)密切相关;Kdm6a与GO:0035097(组蛋白甲基转移酶复合体)、GO:0071557(组蛋白H3-K27去甲基化)、GO:0060070(经典Wnt信号通路)、GO:0001701(子宫内胚胎发育)和GO:0048333(中胚层细胞分化)密切相关;Nr3c1与GO:0031946(糖皮质激素生物合成过程的调控)、GO:0016020(细胞膜)、GO:0005737(细胞质)、GO:0006355(以DNA为模板转录的调控)和GO:0016568(染色质修饰)密切相关;Trp53与GO:0097371(MDM2/MDM4家族蛋白结合的分子功能)、GO:0034103(组织重塑的调控)、GO:0090343(细胞衰老的正调控)、GO:0035033(组蛋白脱乙酰酶的调控活性)、GO:0048568(胚胎器官发育)、GO:0009792(出生或卵孵化时胚胎发育终止)、GO:2000269(成纤维细胞凋亡过程的调控)、GO:0035264(多细胞生物生长过程)和GO:0042981(凋亡过程的调控)密切相关。与腭裂形成相关的通路主要有MAPK、Wnt、TGF-β等信号通路,这些差异位点的功能富集主要集中在生长发育、细胞信号传导、代谢等,推测小鼠腭裂的产生有表观遗传学的参与及调控。而这些通路与前人的研究有许多共通之处,一方面验证了前人的研究,另一方面也为探究腭裂疾病与相关基因突变研究提供新的思路。很多实验已证明腭间充质细胞合成的蛋白多糖是腭支架升高的必要条件[12-14]。围绕这一动态发育过程,后续我们可以从糖蛋白信号通路富集的基因中筛选出与腭裂形成的相关基因,进行鉴定和功能验证。NSCPO的病因是复杂的,涉及遗传和环境因素,推测、鉴定基因和研究相关信号通路将是一项艰巨的任务。
