鸭茅的分子育种
2018-09-19张新全黄琳凯赵欣欣
唐 露,张新全,黄琳凯,张 旭,赵欣欣,聂 刚,许 蕾
(四川农业大学动物科技学院,四川 成都 611130)
鸭茅(Dactylisglomerata)又名鸡脚草或果园草,隶属于禾本科(Gramineae)鸭茅属[1],原产于欧洲、北非和亚洲温带地区,主要分布于欧亚温带和北非,全属仅一个种,即鸭茅[2]。鸭茅作为一种优良的冷季型牧草,具有叶多高产、耐阴、适应性强、适口性好、营养价值高等优点,可用于青饲、调制干草或青贮,是世界四大广泛分布的禾本科牧草之一,全球每年约生产14 000 t鸭茅种子,占世界温带牧草种子的3.3%[3-4]。除此之外,在石漠化治理、退耕还草,建立粮-草、草-草、林-草复合植被等生态工程及草地建设中鸭茅也发挥着重要作用[5]。
随着岩溶石漠化治理和南方现代草地畜牧业推进行动项目的实施,鸭茅作为重要的石漠化治理草种,需求量极大,因此选育出质优、适应性强的鸭茅新品种至关重要。但是,由于鸭茅是多年生异花授粉植物,其遗传杂合性高,许多重要农艺性状属于微效多基因控制的数量性状,遗传机理不明,利用常规育种方法手段难以满足不同目的定向培育鸭茅新品种的要求。利用常规育种结合分子技术可显著提高育种效率,缩短育种周期,加快选育进程[6-7]。与其他重要农作物相比,利用现代先进生物技术进行鸭茅遗传改良虽然起步较晚,但在种质资源的评价、遗传多样性分析、指纹图谱构建、遗传图谱构建、重要性状的基因定位、基因的遗传转化等方面已取得了一些初步的成就。为此,回顾近年来国内外鸭茅分子育种的主要研究进展,并对现代分子生物学技术在鸭茅育种应用中存在的主要问题和应用前景进行讨论和展望,以期供鸭茅育种研究者借鉴。
1 鸭茅品种选育现状
根据国际植物新品种保护联盟(International Union for the Protection of New Varieties of Plants,UPOV,http://www.upov.int/portal/index.html.en)公布的数据,其成员国互认的鸭茅登记品种一百多个,且大多为综合品种。而我国鸭茅育种工作从20世纪80年代起步,截至目前登记的国审品种仅有15个,其中“滇北”、“古蔺”、“宝兴”、“川东”、“滇中”5个为野生驯化品种,“安巴”、“草地瓦纳”、“德娜塔”、“大使”、“波特”、“皇冠”、“阿索斯”、“英都仕”、“阿鲁巴”和“斯巴达”10个为引进品种,主要在我国西南地区推广应用。目前鸭茅品种选育方法较单一,主要为常规育种,利用现有的种质资源进行简单评价筛选后选育而成[8-9],而作物上已较为成熟的分子标记辅助育种、转基因育种等在牧草上应用较少,加快鸭茅分子育种进程,丰富鸭茅品种选育方法,还需广大育种研究者大力推进。
2 遗传多样性分析与指纹图谱构建
2.1 遗传多样性研究
遗传多样性研究能真实反映材料在遗传物质上的差异,明确材料间的亲缘关系,避免了在选配杂交亲本时,因遗传基础狭窄影响选配效率,从而为鸭茅杂交育种及遗传改良提供理论依据。分子标记是进行遗传多样性分析最有效的工具。分子标记种类繁多,性能各异,Costa等[10]对随机扩增多态性DNA标记(random amplified polymorphic DNA,RAPD)、ISSR(inter-simple sequence repeat)和扩增片段长度多态性(amplified fragment length polymorphism,AFLP)3种标记比较研究发现,AFLP是最适合于指纹鉴定和评估鸭茅不同基因型间亲缘关系的标记。Mao等[11]研究表明,简单重复序列(simple sequence repeat,SSR)可以检测到较多遗传位点,可视为研究鸭茅遗传多样性的有效工具。
国内外研究表明,来自不同自然居群的鸭茅其遗传多样性存在差异,同时与其倍性也密切相关。使用SSR和ISSR标记并结合表型对希腊3个不同自然居群的72份鸭茅材料进行了遗传多样性分析结果表明,南部的居群材料比北部具有更高的遗传多样性[12]。Last等[13]分别从瑞士、挪威和保加利亚3个区域的59个自然或半自然居群中收集到1 861份鸭茅材料,利用SSR标记对其倍性、种群遗传多样性及等位基因多样性进行研究,结果表明,3个居群鸭茅材料皆为四倍体,且不同居群间鸭茅遗传多样性和稀有等位基因含量都存在明显差异。倍性方面,Tuna等[14]对土耳其特雷斯地区的57个鸭茅自然居群的倍性和遗传多样性进行研究发现,大部分的鸭茅为四倍体,二倍体居群较少,且四倍体具有更丰富的遗传多样性,聚类分析表明,不同自然居群间有明显的基因交流。彭燕等[15]用AFLP对国内外35份野生鸭茅材料的遗传多样性进行评估发现,二倍体鸭茅和四倍体材料之间虽然亲缘关系较近,但四倍体遗传多样性比二倍体更丰富。万刚等[16]利用SSR标记对23份鸭茅二倍体、四倍体鸭茅研究亦表明,四倍体较二倍体材料的遗传多样性更为丰富。由此可见,四倍体个体是鸭茅品种选育的优良材料,而多倍体则可视为饲料作物选择中一个重要的价值参考因素,以此提高牧草的产量和质量。
鸭茅遗传多样性与海拔、地理分布、管理强度和生长地状况等同样有一定的相关性。Reeves等[17]用AFLP标记研究了法国和意大利的鸭茅居群,发现生长于不同海拔的鸭茅居群间存在遗传多样性。张成林等[18]同样采用AFLP对来自中国新疆和吉尔吉斯坦2个地区90份鸭茅材料进行遗传多样性分析,发现遗传多样性与海拔升高呈显著负相关关系,与年均温增加呈显著的负相关关系,与年降水量、经纬度没有显著的关系。Sun等[19]对来自中亚和中国天山及西南3个不同地理分布的41份鸭茅材料研究发现,不同地理来源的鸭茅遗传多样不同。Jiang等[20]利用起始密码子靶向多态性标记(start codon targeted polymorphism,SCoT)对来自7个不同区域21个国家的95份鸭茅材料进行遗传距离和水平研究,结果发现,来自中国两个区域的材料相似性最高,95份材料根据其地理来源被聚为7类,但也有个别材料在聚类时出现意外。另有其他学者用RFLP(restriction fragment length polymorphism)、RAPD等标记对不同区域的鸭茅自然居群材料的遗传变异情况分析,各居群间的遗传多样性水平存在差异,也存在较高水平的基因交流[21-22]。此外,Boller和Herzog等对瑞士和挪威的半自然鸭茅居群进行遗传多样性研究表明,随着管理强度的减小,导致遗传位点内基因型多样性下降[23-24]。总而言之,鸭茅的遗传差异可能受地理生态环境、气候条件影响,也与育种系统和基因交流有一定相关性,甚至鸟类的迁徙和育种材料的选择都与其相关。而适度放牧有助于基因流发生,也是种群遗传多样性维持的重要机制,为鸭茅属物种遗传保护提供了重要依据。
2.2 DNA指纹图谱在品种鉴定中的应用
传统的植物品种鉴定通过形态学及生育期特性结合的方法进行,但易受环境影响,且存在定义不明确、观测误差大等问题。而DNA指纹图谱是能够鉴别生物个体差异的电泳图谱,直接反映多态性丰富,有高度的个体特异性和环境稳定性,弥补了传统方法的不足[25]。AFLP、SSR、ISSR、RAPD、SRAP、EST-SSR、SCoT等DNA指纹标记作为植物品种鉴定最有效的方法,已在多花黑麦草(Loliummultiflorum)[26]、羊草(L.chinensis)[27]、高羊茅(Festucaelata)[28]、老芒麦(Elymussibiricus)[29]等多种禾草上应用。迄今为止,有关鸭茅在指纹图谱构建的报道,仅见蒋林峰等[30]采用组合引物法,运用4对SSR引物(A01E14、A01k14、B03E14、D02K13)和1个ScoT标记(SCoT23)鉴定我国21个鸭茅主栽品种,结果表明,所有的供试鸭茅品种能够完全区分开,且随着引物组合数量的适当增加,其代表DNA水平信息将更加丰富、全面和准确,具备更强的品种鉴别能力,适于大规模数量的鸭茅品种鉴定。鸭茅指纹图谱构建的研究太少,今后还应继续选择和增加品种鉴定所需核心引物,以便适时扩充新的有效核心引物数据进入指纹图谱数据库,从而保证数据的准确性和实用性。
3 遗传图谱构建及重要性状定位
植物育种实践表明,突破性饲草品种的选育往往取决于重要基因资源的发掘利用。分子标记辅助下的多基因聚合分子育种将是今后突破性饲草选育开拓性研究的领域,分子标记辅助选择技术(marker assisted selection,MAS)为多基因聚合选择提供了强有力的工具[31]。重要农艺性状QTL(quantitative trait loci)定位分析是分子标记辅助选择育种的基础,而高密度遗传图谱构建又是重要农艺性状QTL分析的基础,在遗传图谱的基础上检测数量性状和分子标记的关联性,解析复杂性状和探究表型变异的遗传力,寻找主效QTL紧密连锁的分子标记进行辅助选择育种[32]。
3.1 遗传图谱构建
据不完全统计,国内目前已构建了23余个牧草品种50余张遗传连锁图谱,涉及豆科苜蓿属、三叶草属、百脉根(Lotusccorniculatus),禾本科羊茅属、黑麦草属、鸭茅属和结缕草属等常见草种[33]。有关鸭茅遗传图谱构建研究报道较少,截至目前国内外构建了4张鸭茅遗传图谱(表1)。2011年,Song等[34]首次报道了同源四倍体鸭茅的SSR连锁遗传图谱,其中父本的遗传图谱包含24个连锁群,总长度为562 cM,有168个位点,平均图距3.3 cM,母本包含26个连锁群,总长度为745 cM,有227个位点,平均图距为3.3 cM,亲本图谱间同源连锁群共有7个。Xie等[35-36]则采用SRAP(sequence-related amplified polymorphism)、SSR两种标记构建了世界上首张二倍体鸭茅遗传图谱,并随即采用双拟测交策略构建了包含284个单株的F1代作图群体,使用20对AFLP引物和65对SSR引物构建了另一张同源四倍体鸭茅高密度遗传图谱。
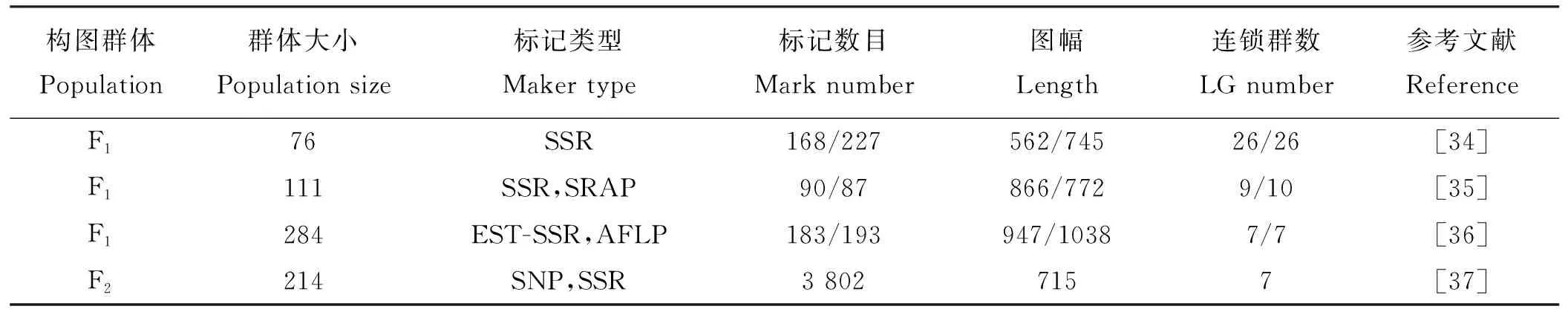
表1 已报道的鸭茅遗传图谱Table 1 Genetic maps of orchardgrass
2016年四川农业大学报道了由2 510个标记构建的总长度715.77 cM,平均图距0.37 cM的高密度四倍体鸭茅遗传图谱,其独特之处在于采用SLAF-seq(specific-locus amplified fragment sequencing)技术和HighMap软件开发了数以万计的高密度分子标签,最终经过分析与筛查将符合要求的SNP标记用于图谱构建。该研究结果极大地推进了鸭茅重要农艺性状QTL定位及功能基因挖掘与研究的进程[37]。
3.2 重要农艺性状定位
许多重要的农艺性状是由多基因控制的,称为数量性状位,受到多种因素的影响,通过构建遗传连锁图谱(genetic linkage map)来对QTL进行定位即连锁作图,也称为QTL作图[33]。鸭茅中利用分子标记进行基因定位、克隆的报道不多,具体性状及方法如表2所列。Xie等[36]通过QTL分析在不同年际间共检测到7个抽穗期相关的QTL,各QTL所解释的表型变异范围为7.85%~24.19%,且将检测到的QTL与近缘物种基因组比较证实,该研究所检测到的QTL是有效的。随后,Zhao等[37]同样通过高密度的SNP遗传图谱对抽穗期及开花期进行QTL分析,4个环境中共检测到11个与目标性状显著相关的QTL,贡献率为8.2%~27%,并找到与抽穗和开花密切相关的候选基因hd1和vrn1。笔者所在的四川农业大学牧草分子育种研究团队目前也正致力于鸭茅生物量相关的重要农艺性状如株高、分蘖、叶长等进行QTL定位研究,力图开发更多与目标性状紧密连锁的分子标记。基于连锁不平衡(linkage disequilibrium,LD)发现新等位基因的方法称为关联分析(association analysis),亦被称为LD作图[42]。Yan等[39]采用EST-SSR和ScoT两种标记对75份抗锈病程度不一的鸭茅进行了遗传多样性分析,并通过关联分析找到了20个与锈病显著关联的分子标记,为鸭茅抗锈病分子标记辅助育种和关联作图分析提供了重要信息。同时Zeng等[40]通过GWAS关联到5 211个与鸭茅锈病相关的SNP标记,并挖掘到两个抗病性候选基因细胞色素P450及醇溶蛋白基因。Zhao等[41]对4个候选基因DgCO1、DgFT1、DgMADS、DgPPD1-like进行了抽穗期与多态性位点关联分析,其中DgCO1含有最多的显著性相关多态性位点且贡献率高,可用于今后鸭茅不同开花期品种选育研究。
4 鸭茅分子标记开发及重要功能基因挖掘
4.1 分子标记开发
比起传统的通过构建基因文库再对其相关区域克隆进行Sanger测序开发分子标记,新一代测序NGS(next generation sequencing)的迅速发展为分子标记的开发带来新的机遇,其耗时少,成本低、准确性也大大提高。Hirata等[43]从1个日本鸭茅品种的4个富含SSR序列的基因组文库中分离得到969个SSR克隆序列,用于引物设计,经验证有676个SSR标记可扩增出条带,其中606个标记显示出多态性。为了丰富鸭茅标记数量,提高鸭茅遗传资源利用率,Bushman等[44]利用双端测序从盐、干旱、和冷胁迫处理的鸭茅组织的cDNA 文库中开发了许多EST序列 (expressed sequence tags, EST)和1 162个EST-SSR标记,并利用水稻(Oryzasativa)、小麦(Triticumaestivum)等同源序对其进行注释。Huang等[45]基于转录组测序从7 764个Unigenes里开发了8 475个EST-SSR 标记,并从75 875个Unigennes鉴定到669 300个高质量的SNP。李季等[46]从基因组Survey 测序中鉴定出78 984 个SSR 位点,并开发出67 216 对Genomic-SSR引物,经验证其扩增成功率高、多态性强且效率高,有望在鸭茅分子育种中发挥重要作用。

表2 鸭茅重要农艺性状定位Table 2 Mapping of main agronomic traits of Orchardgrass
4.2 重要功能基因挖掘
关于鸭茅重要功能基因的挖掘主要通过基因探针捕获、RNA-seq技术等方式挖掘重要功能基因,相关报道如表3所列。Xie[47]通过查询近缘物种开花基因的保守序列,设计基因探针,进行鸭茅基因组开花基因序列的捕获,最终通过序列同源比对找到了与开花相关vrn1、vrn3基因同源序列。Huang等[45]通过RNA-seq技术对两份重要的鸭茅资源“宝兴”(耐热)和“01998”(热敏感)进行研究,通过Illumina HiSeq 2000测序分别得到了3 527(宝兴)及2 649(01998)个差异表达基因,并通qRT-PCR对随机挑选的8个DEG进行了验证,挖掘到大量耐热相关的候选基因,并正在对ZFP、PIP及XTH这3个候选基因进行功能验证研究。Feng等[48]对鸭茅开花前后6个不同时期的RNA-seq分析,发现春化期间有4 689个基因表达明显增加,3 841个基因下降,并挖掘到与春化及花芽发育过程相关的候选基因CCAAT基因家族,并发掘相关转录因子WRKY,NAC,AP2/EREBP,AUX/IAA,MADS-BOX,ABI3/VP1,bHLH,其中MADS-BOX可能参与春化调控。Zou等[49]通过分析处理前、5 h水处理及5 h秋水仙素处理的根尖转录组信息,发现3 381个基因在5 h水处理后进行了差异表达,而3 582个基因在5 h秋水仙素处理后进行了差异表达。比较两个处理的转录组信息,共筛选出2 247个表达显著差异基因,这些基因参与了苯丙素生物合成、苯基丙氨酸代谢、植物激素信号转导、淀粉蔗糖代谢、细胞凋亡、化学致癌等通路。随着高通量测序技术的飞速发展,对功能基因挖掘的方法将更加便捷快速,但如何在数以百万计的测序结果中,进行高效的生物信息学分析,筛选到重要的目标基因,则还需要不断地深入探讨。
基因挖掘、定位的最终目的是克隆目标性状基因并将其应用于育种实践。鸭茅基因克隆及表达分析等工作虽然已经起步,但研究内容还比较零散,系统性差。 Alexandrova和Conger[50]通过表型克隆的方式,分离克隆了两个跟体细胞发育相关的基因DGE1和DGE2。董志等[51]通过RT-PCR技术从鸭茅斑驳病毒(fCMV)总RNA中获得了外被蛋白(CP)基因,转化大肠杆菌后,表达产物经SDS-PAGE和Westernblot分析证明,CP基因在大肠杆菌中获得了高效表达。季杨[52]通过同源克隆的方式,首次克隆了鸭茅中调控抗氧化酶(SOD、CAT和POD)的相关基因。此外,鸭茅基因克隆研究大多关注于抗逆性基因,相关研究工作为鸭茅抗逆育种奠定了重要基础。

表3 已报道的鸭茅重要功能基因挖掘Table 3 Important functional gene mining of Orchardgrass
5 遗传转化体系建立及转基因研究
转基因牧草具有许多优良性状,能提高牧草的质量,还可增强对不利因素的抗性,因此转基因技术在牧草上的运用越来越广泛。目前,牧草中转基因研究在苜蓿(Medicagosativa)、百脉根、高羊茅、白三叶(Trifoliumrepens)上较为成功,例如,抗虫害转基因苜蓿、抗禽流感转基因百脉根等都在我国培育成功[53-54]。而鸭茅由于其遗传背景复杂,高频遗传转化体系难以构建,因此转基因研究报道较少,相关研究如表4所列。最早关于鸭茅遗传转化体系建立的报道见于1972年,Schenk和Hildebrandt[55]将鸭茅叶片沿中轴撕开,横切成3 mm小段后于SH 基础培养基上培养即可得到愈伤组织。1982年,Conger等[56]先将 ‘Potomac’鸭茅最内部幼嫩叶片诱导3~4周后转入SH继续培养得到鸭茅再生植株,然后发现,使用鸭茅叶肉细胞培养,可以不产生愈伤而直接发生体细胞胚。1984年Gray等[65]提出和建立了鸭茅悬浮细胞系,并且成功诱导胚性愈伤的形成。在悬浮培养基中利用鸭茅幼嫩叶片培育形成胚状愈伤组织,这些悬浮液即被用来作为原生质体,用于遗传转化。随后通过电热击法和PEG法,成功将抗潮酶素基因转入该鸭茅原生质体系中,经过潮霉素选择培养基筛选和对转化子Southern杂交鉴定后,由胚形愈伤诱导而成的转基因植株含有潮霉素抗性基因[57-58]。1997年Denchev等[59]通过基因枪法将包含bar和GUS(报告基因)基因的DNA微弹直接轰击转入鸭茅叶肉细胞形成的胚形愈伤组织中,共转化率达到30%。2000年Lee等[60]通过同样的方式,首次通过农杆菌介导转化得到两个品种的鸭茅转基因植株,从而改善了以往遗传转化率相对较低的现状。2001年,他们将BcGR1(谷胱甘肽还原酶)基因转入鸭茅植株中[61]。同年,Nakumar等[62]则利用四倍体鸭茅种子建立了一个高频遗传转化体系,并将两个抗2,4-D基因转入愈伤组织,共转化率达30%,得到的T0代植株倍性变化极大,30%为四倍体,70%为八倍体。Kim等[63]在2005年将一个耐热基因DgP23通过农杆菌转入鸭茅愈伤组织,且将转化而得的植株置于X射线中处理,但得到的转基因植株在表型上与野生型却没有任何差异。并在2008年将另一个耐热基因DgHsp17.2转入鸭茅,虽然得到的转基因植株在田间及温室试验与野生型没有太大差异,但将转基因植株置于致死温度时,表现出较强的耐热性[64]。Lee[66]2003年开始研究鸭茅品种及各种生长调节剂对构建高频转化体系的影响,并在2006年利用鸭茅种子建立的遗传转化体系,继续探讨农杆菌转导中如何提高其遗传转化率[67]。但之后近10年关于鸭茅转基因研究均未见报道,鸭茅作为重要的冷季型禾草,其基因功能分析研究方面几乎处于刚刚起步的阶段,如何完善鸭茅再生及高效遗传转化体系,深入重要目标基因的功能研究也是笔者所在四川农业大学牧草分子育种研究团队近年来正在全力攻克的一大难题。
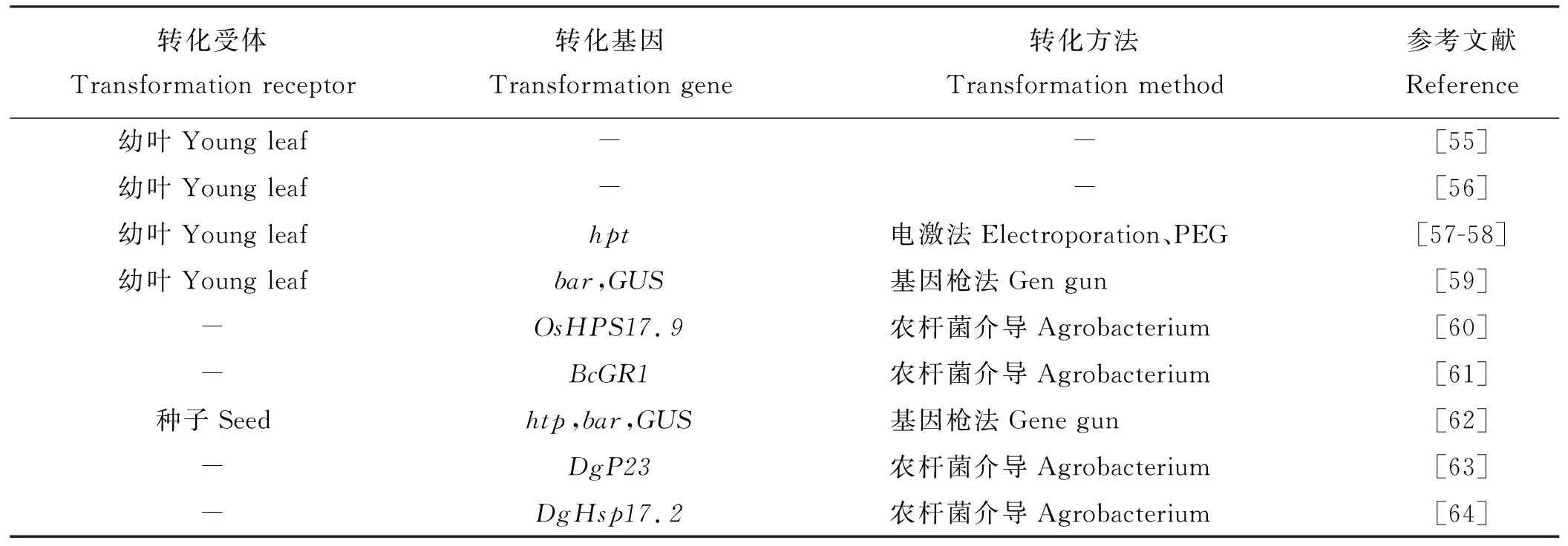
表4 已报道鸭茅转基因相关研究Table 4 Transgenic studies in Orchardgrass
6 研究展望
综上所述,相较于其他重要经济作物,鸭茅分子育种工作起步较晚,但在遗传多样性分析、遗传图谱构建及转基因研究等方面取得了较大的进展,可见分子育种技术对于加快育种进程、提高选择效率及预见性方面是可行的,但目前尚存一些问题仍待进一步探讨。笔者认为,就鸭茅的分子育种现状而言,未来应不断推进以下几方面工作:
1)基因组学的发展将为分子育种带来全新的发展契机。由于大多数牧草是多年生异花授粉植物,其遗传杂合性高,基因组结构复杂,全基因组测序存在一定难度,目前仅有蒺藜苜蓿(M.truncatula)[68]、红三叶(Trifoliumpratense)[69]、二穗短柄(Brachypodiumdistachyon)[70]、多年生黑麦草(L.perenne)[71]、日本百脉根(L.japonicus)[72]、柳枝稷(Panicumvirgatum)、狼尾草(Pennisetumalopecuroides)[73]、狗尾草(Setariaitalica)[74]完成全基因组测序。四川农业大学牧草分子育种研究团队通过对鸭茅的基因组进行基本信息调查之后,随即开展测序,已完成全部序列测定、拼接及功能注释,届时对解析鸭茅基因组结构及特征、亲缘关系及遗传进化、功能基因组学、蛋白质组学及生物信息学等的发展提供海量信息,也可对其他禾本科牧草育种提供借鉴。
2)分子标记辅助选择育种将显著提高育种效率。重要农艺性状相关基因的定位和分子标记的开发大量工作正在实施,通过连锁及关联分析等定位抗逆、品种和产量等重要农艺性状相关基因位点,阐明优良种质资源中携带的优异等位变异,开发育种可用的理想分子标记,为分子标记辅助育种奠定基础。
3)多基因聚合育种为快速选育高产优质高抗的综合品种提供更多可能。过去常用的杂交、回交、复交等传统育种手段,过程繁杂缓慢,聚合效果不理想。而植物多基因聚合育种可快速准确地聚合植物的优良性状,将定位或挖掘的与鸭茅目标性状紧密连锁地功能分子标记或候选基因进一步研究后,结合全基因组选择及新一代基因编辑技术,如CRISPR/Cas,通过分子标记辅助选择和遗传转化等多基因聚合育种方法,对牧草育种进行定向设计。尤其是CRISPR/Cas9系统能够实现对基因组的特异性和精确敲除,且易于操作、成本低、构建周期短并且可以实现同时对多个基因的编辑,在苜蓿、高粱、水稻、玉米已经取得突破,相关成功经验可为鸭茅分子聚合育种提供借鉴。
