不同营养因子对土壤细菌可培养物种多样性的影响
2018-09-11景凤霞李云琪唐灵杰李小锦王宇辉张秀敏
景凤霞,李云琪,唐灵杰,李小锦,王宇辉,张秀敏
(河北大学生命科学学院,河北 保定 071002)
自然生境中的微生物物种非常丰富,但可培养物种仅占环境微生物总数的0.1%~10%[1]。大多数对人类有益的微生物仍处于一种不可培养的状态,难以得到其纯培养物。传统的培养方法是向样品提供丰富的营养物质,然后选择快速生长的菌落,这些培养方法仅能获得自然生境中的一小部分细菌[2],这就使得绝大多数细菌难以被研究和利用。许多研究者通过改变培养基成分和培养条件或创新培养方法来解决这一问题。岳秀娟等[3]在培养基中添加丙酮酸钠、过氧化氢酶和甜菜碱等物质用以培养微生物,这些物质可以减少微生物代谢过程中产生的活性氧,结果显示添加活性氧清除能力物质的平板上菌落数比对照组明显增加,并且分离得到多株具有抗菌活性的微生物,这种方法为抗生素的筛选和菌种的保藏提供了新的来源。高丝氨酸内酯作为一种信号分子也被普遍研究[4]。
Shayne等[5]设计了以VL55培养基为基础培养基,向培养基中加入不同的生长因子,共分离得到包括9门17纲60科的350株细菌,序列比对结果显示,有93株(27%)细菌为新属。Tanaka等[6]在灭菌前将磷酸盐和琼脂分开,菌落生长的数量增多,其中超过30%属于未培养微生物。目前,越来越多的科研工作者致力于利用宏基因组分析方法来寻找新的未培养微生物,研究其遗传多样性和微生物群落的结构,以及了解其在生态系统中的重要作用[7-8],并通过使用16S rRNA基因序列对未培养的细菌进行多样性分类[9]。也有一些学者通过采用富集培养的方法,获得之前未培养的微生物[10]。
本试验以改良的VL55为基础培养基,向其中添加6种不同的营养因子,通过大量稀释对采自河北、青海和云南的3份土壤样品进行细菌分离培养,比较可培养细菌的物种多样性情况,以期发现更多新的微生物类群,为开发新材料、新药物提供丰富的微生物物种资源。

图1 土样1分离菌株的多样性分布

图2 土样2分离菌株的多样性分布

图3 土样3分离菌株的多样性分布
1 材料与方法
1.1 试验材料
土样采集:试验于2016年在河北省微生物多样性研究与应用实验室进行,供试土壤样品分别采自河北涞水县上港村、青海三江源、云南无量山3个地区,分别记为土样1、土样2和土样3,分别风干、研碎、除去石子和细根,过2 mm筛备用。
培养基:改良的VL55基础培养基:2-(N-吗啉)乙磺酸3.9 g,七水合硫酸镁0.048 g,氯化钙0.067 g,磷酸氢二铵0.053 g,丙酮酸钠0.8 g,结冷胶3%,蒸馏水1 L。灭菌前加入2 mL亚硒酸-钨酸盐溶液[11]和2 mL 微量元素溶液SL-10[12]。 用 0.2 mol/L 的 NaOH 和 0.1 mol/L 的KOH混合液调节改良的VL55基础培养基pH与土壤pH一致,高温灭菌后,在使用前加入维生素溶液1和维生素溶液2[13]。然后分别添加6种不同的营养因子(表1)。
PCR扩增试剂:使用细菌通用16S rRNA基因引物27f (5’-AGAGTTTGATCCTGGCTCAG-3’)[15]和1492r(5’-GGTTACCTTGTTACGACTT-3’)[16],引物由通用生物系统(安徽)有限公司合成。PCR反应体系:TaqDNA聚合酶、10×Taq PCR Buffer(含Mg2+)、10 mmol /L 高纯dNTPs(北京康为世纪生物科技有限公司)。细菌基因组DNA提取试剂盒,北京康为世纪生物科技有限公司。

表1 6种不同营养因子组合
1.2 试验方法
1.2.1 细菌分离与保存 土样1、土样2和土样3各称取1 g样品,分别加入99 mL VL55液体培养基中,振荡混匀,取0.5 mL土壤悬浮液于4.5 mL试管中进行10-1~10-7梯度稀释,之后涂布分离进行细胞计数,根据计数结果确定100 μL菌液中尽可能低于1个细胞的稀释度。经统计得出土样1的10-4梯度每100 μL菌液中含细胞总数为5个,土样2的10-4梯度每100 μL菌液中含细胞总数为5个,土样3的10-5梯度每100 μL菌液中含细胞总数为3个。因此,3种土壤样品均选择10-4、10-5、10-6、10-74个梯度进行大量稀释,每个梯度取100 μL至装有5 mL添加不同营养因子组合的VL55液体培养基的试管中,直至将每个梯度试管内的液体分装完毕,每个梯度分装45支试管,每种土样稀释液共分装试管1 080支。然后置于28℃、200 r/min恒温摇床培养。
待试管内的培养液浑浊后,接种至R2A固体平板上,置于28℃生化培养箱中倒置培养;待平板上长出菌落后,挑取单菌落于R2A液体培养基中,置于28℃、200 r/min摇床振荡培养;待菌体长出后,用显微镜验纯,吸取0.5 mL菌液于含有50%甘油的甘油管中,每株菌保藏两支甘油管,-20℃保藏菌体。剩余菌体经12 000 r/min离心、收集,用于提取基因组DNA。
1.2.2 细菌DNA的提取和16S rRNA基因扩增 使用康为世纪生物科技有限公司的细菌基因组DNA提取试剂盒提取菌株的基因组DNA。使用细菌通用引物27f和1492r对菌株的16S rRNA基因进行PCR扩增。
PCR扩增体系为50 μL,其中包括0.5μL上游和下游引物(20 μmol/L),2 μL DNA(70 ng/μL)模板,1 μL dNTP (10 mmol/L),5 μL 10×Buffer,0.3 μLTaqDNA 聚合酶(5 U/μL),加入无菌去离子水至要求的体积。PCR扩增条件:94℃预变性5 min;94℃变性1 min、55℃退火45 s、72℃延伸1 min,30个循环;72℃延伸10min。
1.2.3 16S rRNA基因测序及系统进化分析 扩增产物经1%琼脂糖凝胶电泳检测并回收,送通用生物系统(安徽)有限公司测序。利用EzBiocloud对菌株测序所得的16S rRNA基因序列进行同源性比对,找出亲缘关系最近的模式菌株的16S rRNA基因序列及相似值。再利用BioEidt和MEGA 5.0软件对分离得到的新物种进行系统发育分析,系统发育树的构建采用Neighbour-Joining模型,进而确定其系统发育地位。
2 结果与分析
2.1 土壤细菌菌株分离结果
2.1.1 土样1 将土样1进行梯度稀释,再经大量稀释分装于添加不同营养因子的VL55液体培养基中,将分离所得菌株的16S rRNA基因序列在EzBioCloud中进行同源性比对,得出土样1的分离结果,对照分离得到5个属,添加6种营养因子的处理共分离得到22个属。从图1(封三)可以看出,土样1添加6种不同营养因子处理均分离到对照未分离到的微球菌属(Micrococcus)的菌株,其中,添加营养因子组合1分离得到特有的农球菌属(Agrococcus)、考克氏菌属(Kocuria)和塚村氏菌属(Tsukamurella)的菌株,添加营养因子组合2分离得到特有的短杆菌属(Brevibacterium)、纤维单胞菌属(Cellulomonas)、肠杆菌属(Enterobacter)和原小单孢菌属(Promicromonospora)的菌株,添加营养因子组合6分离得到特有的博斯氏菌属(Bosea)、极单胞菌属(Polaromonas)和鞘氨醇杆菌属(Sphingobacterium)的菌株。
2.1.2 土样2 将土样2按照土样1的处理方式进行处理,其分离结果(图2,封三)显示,对照分离得到2个属,添加6种营养因子的处理共分离得到17个属,其中,处理1分离得到特有的鞘氨醇杆菌属(Sphingobacterium)的菌株,处理2分离得到特有的慢生根瘤菌属(Bradyrhizobium)、东属(Dongia)和贪噬菌属(Variovorax)的菌株,处理3分离得到特有的阿菲波菌属(Af i pia)的菌株,处理5分离得到特有的放线多形菌属(Actinopolymorpha)、卡西列罗属(Caballeronia)和莫拉氏菌属(Moraxella)的菌株,处理6分离得到特有的类芽孢杆菌属(Paenibacillus)的菌株。
2.1.3 土样3 将土样3按照土样1处理方式进行处理,其分离结果(图3,封三)显示,对照分离得到6个属,添加6种营养因子的处理共分离得到15个属,其中,处理1分离得到特有的无色小杆菌属(Achromobacter)和根瘤菌属(Rhizobium)的菌株,处理2分离得到特有的贪铜菌属(Cupriavidus)和贪噬菌属(Variovorax)的菌株,处理5分离得到特有的假芽孢杆菌属(Fictibacillus)的菌株,处理6分离得到特有的节杆菌属(Arthrobacter)和纤维单胞菌属(Cellulomonas)的菌株。
2.2 潜在新种16S rRNA基因相似性比对结果
目前认为划分细菌新种的16S rRNA基因序列相似性阈值是98.65%[17]。通过在改良的VL55培养基中添加6种不同的营养因子,利用大量稀释法对采自河北、青海和云南的3份土壤样品进行微生物物种的分离。结果(表2)显示,有12个菌株与其参比菌株的16S rRNA基因序列相似性均低于98.6%。其中,土样1未分离得到序列相似性低于98.65%的菌株,不存在替在新种,土样2、土样3分别分离得到4株和8株潜在新种。因此,这12个菌株很有可能为潜在的新种,按隶属7个不同的属分别构建系统发育树,结果见图4~图9。

表2 不同土样添加营养因子处理分离得到潜在新种的比对结果

图4 依据16S rRNA 基因序列构建的HBUM200206相关菌株的N-J系统发育树

图5 依据16S rRNA 基因序列构建的HBUM200202相关菌株的N-J系统发育树

图6 依据16S rRNA 基因序列构建的叶杆菌属相关菌株的N-J系统发育树
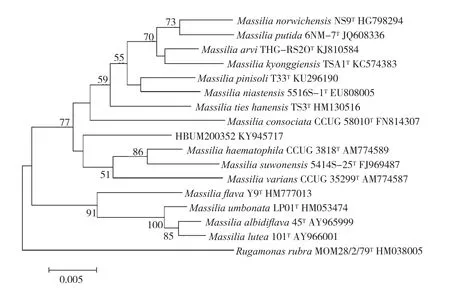
图7 依据16S rRNA 基因序列构建的HBUM200352相关菌株的N-J系统发育树

图8 依据16S rRNA 基因序列构建的副伯克霍尔德氏菌属相关菌株的N-J系统发育树

图9 依据16S rRNA 基因序列构建的HBUM200497相关菌株的N-J系统发育树
3 结论与讨论
本研究通过在培养基中添加不同营养因子,对采自河北、青海和云南的3份土壤样品的微生物进行大量稀释分离,结果表明分离得到的微生物物种多样性均较对照丰富;添加不同营养因子的培养基处理分离得到各自独特的微生物类群,表明不同的营养因子能够选择性分离得到不同微生物类群,对于丰富微生物物种多样性有重要意义。
综合来看,3份土壤样品在添加营养因子1和2的培养基中分离得到的菌株种类均较对照丰富,添加营养因子4的培养基分离得到的菌株种类均较对照少。其中,营养因子1中的N-乙酰葡萄糖胺能够与N-乙酰胞壁酸经β-1,4糖苷键连接间隔排列形成细菌细胞壁的主要成分肽聚糖,添加N-乙酰葡萄糖胺有利于保持细胞外形并保护细菌抵抗低渗环境,起到一定的屏障作用,更有利于细菌存活。
营养因子2中葡萄糖、乳糖、麦芽糖、阿拉伯糖等作为菌株的碳源可以促进其生长繁殖。当培养基中同时含有葡萄糖、乳糖、麦芽糖、阿拉伯糖及其他碳源时,细菌会优先使用葡萄糖作为碳源,而当其耗尽时,细菌需要经过一定时间的适应,才能利用乳糖及其他碳源继续生长。补充不同的碳源会引起菌体生理及生长特性变化,使其分离得到的菌株更为丰富。
营养因子4中乙酸钠、苯甲酸钠、L-乳酸钠均为常见的防腐剂,能够抑制微生物的生长繁殖。乙酸钠作为一种广谱型防腐剂,能够抑制细菌、霉菌及酵母菌的生长;苯甲酸钠具有较强的抑菌作用,可通过扩散的方式被细菌所摄取,改变细胞膜的通透性,抑制细胞膜对氨基酸的吸收;L-乳酸钠对致病菌有很好的抑制作用,因此添加营养因子4使菌株的丰富性下降。
此前,16S rRNA基因序列和DNA-DNA杂交比较的结果显示,70%的DNA-DNA匹配性对应于约97%的16S rRNA基因相似性,即界定种的阈值为97%。Kim等[17]研究报道,区分两个不同种的16S rRNA基因序列相似性的临界值为98.65%,即两株菌的16S rRNA基因序列相似性在98.65%以下,则认为它们属于不同种,不需要做DNA-DNA杂交实验。
本研究结果表明,分离得到的菌株HBUM200206与 运 动 东 菌(Dongia mobilis)LM22T的亲缘关系最近,序列相似性为94.58%,可能是红螺菌科(Rhodospirillaceae)的一个潜在新属。菌株HBUM200202、HBUM200206、HBUM200239、HBUM200240、HBUM200352、HBUM200424、HBUM200427、HBUM200448、HBUM200452、HBUM200488、HBUM200497和HBUM200503与亲缘关系最近的模式菌株的16S rRNA基因序列相似性均小于98.65%,可能为潜在的新种。
