2013年贵州省急性弛缓性麻痹病例中非脊髓灰质炎肠道病毒的鉴定分析
2018-09-04王寅寅唐小敏叶绪芳
王 宇,任 刚,王寅寅,苏 飞,唐小敏,叶绪芳
人类肠道病毒(huamn enteroviruses,HEVs)属于小RNA病毒科肠道病毒属的成员,HEVs是人类普遍的致病病毒之一,目前根据病毒株的基因特征及生物学遗传特性分析已知肠道病毒超过100多种型别,并可分成肠道病毒A、B、C和D 4组[1-2].肠道病毒主要包括脊髓灰质炎病毒(poliovirus,PV)、柯萨奇病毒(coxsackievirus,CV)、埃可病毒(echovirus,ECHO)以及新型肠道病毒(newer enteroviruses),HEVs的主要传染源为病人或无症状带毒者,其可引起多种疾病,特别是引起儿童的疱疹性咽峡炎、手足口病、急性出血性结膜炎、心肌炎和神经系统疾病等亦成为严重的公共卫生问题[3].
急性弛缓性麻痹(Acute Flaccid Paralysis,AFP)系统监测对象主要针对15岁以下儿童出现急性弛缓性麻痹症状的病例,是全球消除脊髓灰质炎(脊灰)和维持无脊灰状态的重要工作组成,但伴随着全球野脊灰病毒的逐渐消灭由非脊灰肠道病毒(non-polio enterovirus,NPEV)引起的麻痹病例引起人们重视.1999年Oberste等[4]提出了基于肠道病毒VP1区序列的分子生物学定型方法,加速了对非脊灰肠道病毒分型及基因特征的研究工作.贵州省自1994年后未发现本土脊灰野病毒病例[5],但每年都会在AFP病例的粪便标本中分离到大量的NPEV,为了解贵州地区NPEV毒株的血清型别特征,依据VP1区基因定型原理,本研究对2013年从贵州省AFP病例粪便标本中分离到NPEV进行了回顾性检测分析,并分析CVA4的VP1基因特征,为今后对NPEV所致相关疾病的防控提供科学依据.
1 材料与方法
1.1 标本与资料来源 肠道病毒分离物来源于贵州省2013年AFP病例监测系统报告的AFP病例的粪便标本,粪便标本由各县区疾病预防控制中心采集,冷藏运送至省级脊灰实验室.粪便标本均按照WHO《脊髓灰质炎实验室手册》第四版[6]的方法用RD(人横纹肌肉瘤细胞)、L20B(转人脊灰病毒受体的小鼠肺细胞)细胞进行培养及病毒分离,收集盲传两代后RD细胞病变而L20B不变的阳性分离物判定为非脊灰肠道病毒(NPEV),将毒株置于-20℃保存.本研究所用的肠道病毒各基因型代表株的VP1基因序列均来源于美国国家生物技术信息中心网站(NCBI).
1.2 病毒RNA提取 采用德国QIAGEN公司的QIAamp Viral RNA MiniKit试剂盒,根据试剂盒的操作说明从病毒分离物中提取病毒RNA.向每管病毒RNA中加入1μL RNA酶抑制剂后-80℃保存备用.
1.3 VP1区RT-PCR及核苷酸序列测定 采用一步法Access QuickTMRT-PCR System试剂盒(美国Promega公司产品)进行(reverse transcript polymerase chain reaction,RT-PCR)扩增NPEV毒株VP1基因.cDNA合成、RT-PCR反应及测序中所用引物参照Oberste等[4,7]的方法进行,PCR反应体系:2×反应缓冲液25μL,AMV酶混合物1μL,上下游引物各1μL,模板RNA 5μL,补足无RNA酶水至总体积50μL.反应条件为45℃30 min,94℃2 min,94℃30 s,55℃30 s,72℃60 s,35个循环后72℃延伸10 min.PCR产物行1.5%琼脂糖凝胶电泳鉴定,对阳性PCR产物进行切胶回收后测序分析.
1.4 基因序列分析和系统进化树构建 采用Sequencher软件对VP1区核苷酸进行拼接整理,通过肠道病毒分子定型数据库Enterovirus Genotyping Tool Version 0.1对序列定型分析;从GenBank数据库中下载肠道病毒各基因型参考毒株VP1区序列,利用MEGA7.0软件通过邻位相接法(neighbor joining method,NJ)构建系统进化树,可靠性通过1 000 bootstrap值评估.
2 结 果
2.1 病毒分离与鉴定 2013年共采集235例AFP病例粪便标本,共分离到25株肠道病毒,其中NPEV为24株,分离率约为10.2%(24/235).另外1株为PV病毒并送国家脊灰实验室进行型内鉴定为PVⅡ型疫苗株,未发现脊灰野病毒.
2.2 NPEV的测序及分子定型 通过RT-PCR对NPEV毒株进行扩增定型,结果见图1.根据Oberste等[4]提出的基于肠道病毒VP1区核苷酸同源性大于75%对病毒株进行型别鉴定,本研究24株NPEV毒株中,有2株无法扩增定型,另外22株共包括12个血清型,其中人类肠道病毒A组包括4个血清型6株,其中3株为CVA4,15株人类肠道病毒B组包括7个血清型,1株CVA21为人类肠道病毒C组,未分离到人类肠道病毒D组病毒.各分离株与其对应原型株的核苷酸(nucleotide,nt)一致性在75.5%~86.8%之间、氨基酸(amino acid,aa)一致性在88.0%~96.7%之间.本次分离的NPEV毒株编号、病毒型别、与对应原型株毒株之间的nt和aa一致性性见表1.
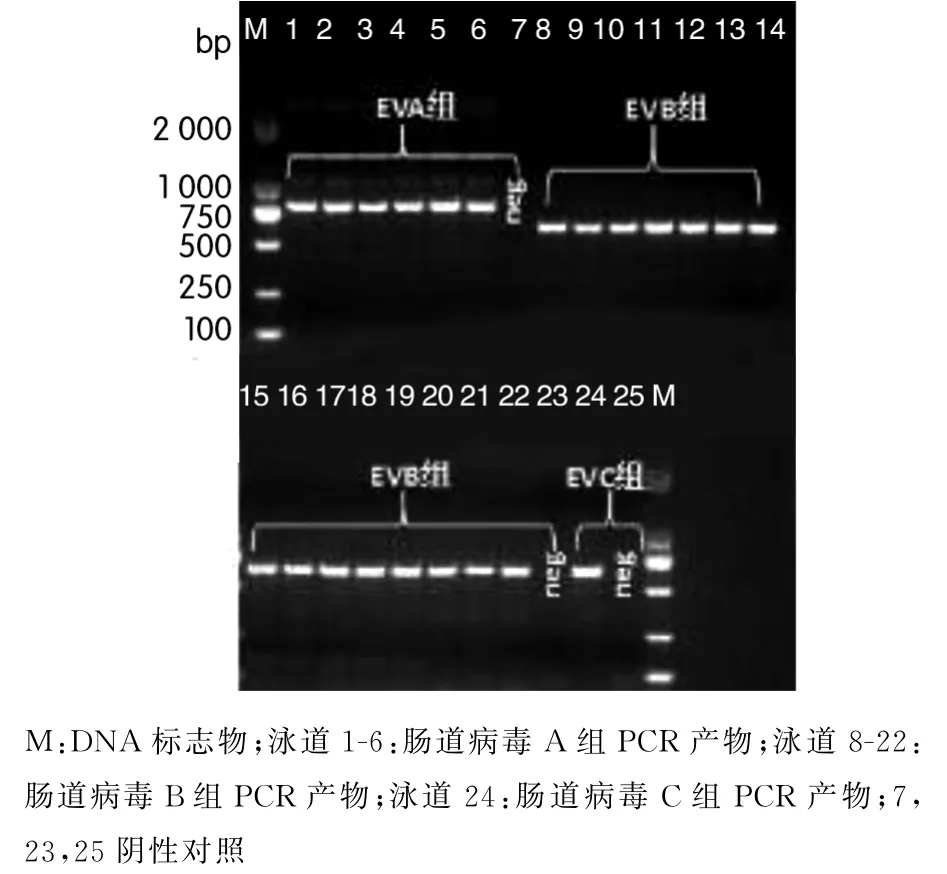
图1 对NPEV毒株进行RT-PCR扩增定型Fig.1 RT-PCR amplification of NPEV strains for genotyping

表1 贵州省2013年分离到的22株NPEV的基本信息Tab.1 Basic information of 22 NPEV isolates in Guizhou Province in 2013
2.3 系统进化树分析 通过GenBank下载已鉴定到的肠道病毒各血清型原型株,22株NPEV贵州分离株与对应原型株之间的亲源关系见图2,相同型别的分离株与对应的原型株聚集在一起,但ECHO7贵州株与其原型株之间进化相对较远.
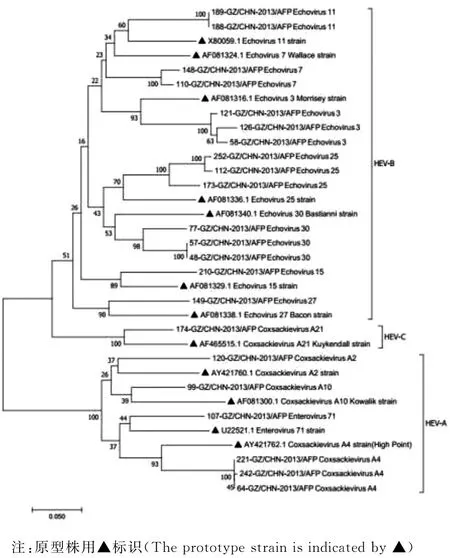
图2 本研究中的22株NPEV与对应原型株之间的基因进化发育树Fig.2 Phylogenetic tree of 22 strains of NPEV and their corresponding prototype strains
2.4 CVA4贵州分离株与其各亚型代表株之间VP1区序列对比分析 本研究分离到3株CVA4,分离率为12.5%(3/24).将3株CVA4贵州分离株与从GenBank中下载的CVA4原型株High point(AY421762)以及CVA4各基因型参考株VP1序列进行平均遗传距离分析见表2,发现CVA4贵州分离株序列与原型株A基因型High point之间的核苷酸平均遗传距离较远,可达18.4%;与CVA4的B基因型代表株Kenya(GQ176232.1)之间的核苷酸平均遗传距离最大达20.6%.通过多重序列比对并构建遗传进化树见图3,发现3株CVA4贵州分离株VP1区之间的核苷酸与氨基酸序列几乎一致,推测可能来源于同一传播链,并与CVA4的C2基亚因型聚为一支,提示CVA4贵州地方株为C2基因亚型.
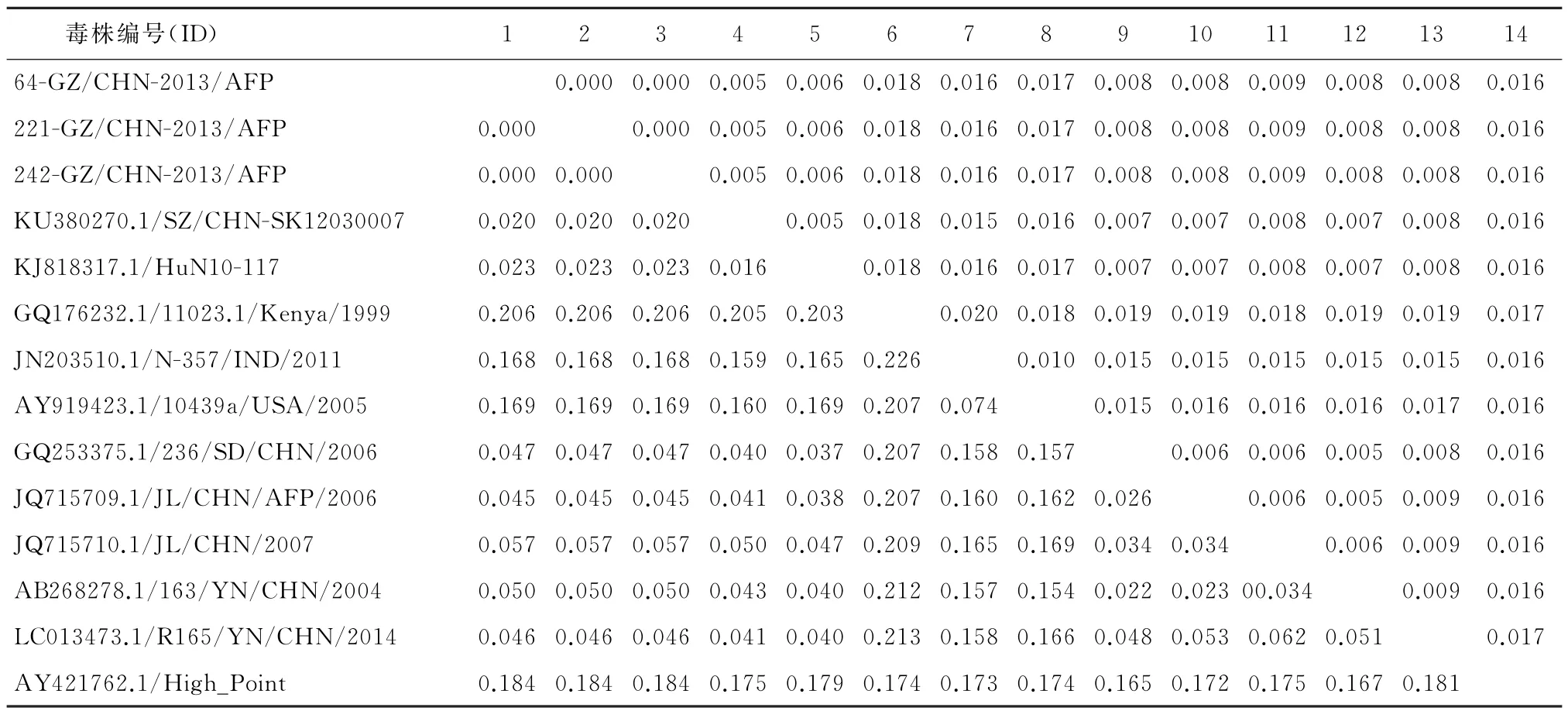
表2 本次分离的CVA4与其各基因型代表株的nt和aa差异率Tab.2 Mean genetic distances of CVA4 strains with strains of different subgenotypes

图3 本研究中分离的3株CVA4与其各基因亚型代表株基因进化树Fig.3 Phylogenetic tree of 3 strains of CVA4 and their corresponding representative subprototype strains
3 讨 论
肠道病毒是人类普遍易感且分布广泛的致病原,随着近年来肠道病毒导致的手足口病在全球的暴发流行越来越受到人们的重视[8].1999年Oberste等[4]提出了基于肠道病毒VP1区序列的分子定型方法,使得以前不能被血清学鉴定的肠道病毒得以发现.VP1基因是主要决定肠道病毒抗原性,具有与血清型完全对应的遗传多样性,基于该技术的运用近年来国内外多个地区相继报道了肠道病毒的分子流行病学特征.国内如山东省[9]和云南省[10]等,国外如印度[11]、尼日利亚[12]和巴基斯坦[13]等,而贵州地区对肠道病毒所致疾病的流行病学特征、临床症状表现以及分子流行病学方面的研究较少,因此本研究对贵州省2013年从AFP病例标本中分离到的NPEV进行分型鉴定,了解贵州省肠道病毒的病原谱及分离到的CVA4的基因特征.
研究发现,2013年贵州省AFP病例中的NPEV以 HEV-B为主,其次是 HEV-A、HEV-C组,没有HEV-D组毒株,研究结果与陈苏等[14]对云南省2015年的NPEV毒株鉴定结果相似,均以HEV-B组毒株为主,提示我国西南地区B组肠道病毒可能分布广泛.本研究中各分离株与其对应原型株的核苷酸一致性在75.5%~86.8%之间、氨基酸一致性在88.0%~96.7%之间,通过构建进化树分析发现各分离株与对应的原型株聚集在一起,但ECHO7贵州株与其原型株之间进化相对较远,提示ECHO7贵州株可能与原型株之间的VP1区存在较大的变异,也可能与肠道病毒B组其它毒株存在重组,值得进一步关注研究.
目前研究证明肠道病毒中有66个血清型的病毒与人类疾病有关联[15],但其是否与引起麻痹有关联还待研究.但近年来人类肠道病毒中除引起手足口病的病原EV71和CVA16的研究报道较多外,CVA4可引发手足口病、疱疹性咽峡炎和急性弛缓性麻痹等疾病也引起广泛关注[16].本研究中分离到的3株CVA4均属于C2基因亚型,与我国陕西、吉林和云南等大多数地区对CVA4的分析类似[17],说明中国大陆CVA4均属于C2基因亚型为优势基因亚型并存在共同进化.目前CVA4尽管没在贵州地区暴发流行,但考虑到在贵州省手足口病聚集性疫情病原谱监测中发现属于非EV71、非CVA16的肠道病毒致病原最多,占32.20%[18],加之肠道病毒大多数为无症状感染状态,提示在实验室中应加强包括CVA4等其他肠道病毒的监测力度,进一步完善区域性肠道病毒流行特征与规律分析,明确毒株之间的遗传进化关系与演替趋势,对制定肠道病毒所致疾病的防控策略提供依据.
