以慢性肾功能衰竭为突出表现的新发致病突变非典型Bardet-Biedl综合征1例报告
2018-08-30申金凤帅兰军吴小川
申金凤,曹 艳,帅兰军,吴小川
中南大学湘雅二医院儿童医学中心,长沙 410011
1 病例资料
1.1 首次入院情况 患儿男性,11岁,因“多饮、多尿2年余,乏力半月余”于2015年3月31日第1次入住我科。患儿2013年上半年无明显诱因出现多饮、多尿,每天饮水约2 200 mL,尿量达3 600 mL/d,伴乏力。于当地医院查肾功能:尿素氮19.12 mmol/L、肌酐440.5 μmol/L;电解质检查:钾1.86 mmol/L;血常规:血红蛋白76 g/L。肾脏彩超示:右肾囊肿。当地医院初步诊断为“慢性肾炎”。既往史、个人史、家族史无特殊。入院查体:身高130 cm(低于134.5 cm两个标准差),体质量32 kg,体质指数(BMI)18.9 kg/m2。发育落后,贫血面容,双眼斜视;手指短,无杵状指及多指/趾(图1);睾丸容积约2 mL,阴茎短小。病理征阴性。入院后查血常规:血红蛋白96 g/L;尿常规:尿蛋白(+)、尿糖()、尿隐血();肾功能:尿素氮21.06 mmol/L,肌酐389.3 μmol/L,肌酐清除率16.36 μmol/L(CKD 4期)。肾脏彩超:双肾实质弥漫性病变;肾脏MRI:双肾多发小囊肿。
肾组织活检:肾组织免疫荧光染色结果IgA(-)、IgG(+)、IgM(+/)、C1q()、C3(+)、C4(-)、Fb(+)。病理检查结果(图2):(1)C1q肾病(增生硬化型);(2)间质纤维化。电镜检查结果:肾小球中度系膜增生,肾小管坏死或萎缩,间质纤维增生。予泼尼松1 mg/kg口服治疗,辅以补钙、控制血压等对症治疗,并于2015年4月19日带药出院。出院时患儿尿素氮25.58 mmol/L,血清肌酐445.5 μmol/L,尿量约2 100 mL/d。出院后规律服药2个月后,自行停药,予中药(具体不详)治疗,病情逐渐加重。

图1 患儿指(A)、趾(B)表现
1.2 再次入院情况 2016年1月28日复查肾功能:尿素氮48.02 mmol/L,肌酐868 μmol/L。患儿伴发热、咳嗽,腹胀,尿少,视物模糊、斜视,偶有胸闷、心悸等不适,遂再次入住我科。入院查体:身高131 cm(低于134.5 cm两个标准差),体质量37 kg,BMI 21.56 kg/m2。身材矮小,双眼斜视,双眼睑轻度水肿,口腔溃疡,伸舌居中、轻微颤动,咽红,扁桃体Ⅱ°肿大,双下肺呼吸音减弱,心律齐、无杂音,腹部膨隆、移动性浊音阳性。睾丸容积约2 mL,阴茎短小,手指短,双下肢中度水肿。入院尿常规:尿蛋白(),尿葡萄糖(+);尿素氮71.73 mmol/L,肌酐1 207.5 μmol/L(肌酐清除率5.27 μmol/L,CKD 5期);甲状旁腺激素140.8 pmol/L。胸部X线:双侧中量胸腔积液,双肺膨胀不全并有渗出性病变。泌尿系彩超:双肾实质弥漫性病变,大小约84 mm×36 mm(左)、77 mm×32 mm(右),右肾体积偏小。心脏彩超:左房增大,直径34 mm,二尖瓣、三尖瓣轻度反流,心包积液。

图2 患儿肾活检组织病理学检测结果
A:右肾穿刺活检组织PAS染色显示,肾小球球周纤维化并肿胀,球内细胞数稍增多,肾小球系膜细胞增生,系膜基质增多,可见轻度渗出与炎性细胞浸润;B:右肾穿刺活检组织PASM染色,肾间质广泛纤维化;C:肾组织免疫荧光染色C1q().Original magnification: ×400(A), ×200(B), ×100(C)
采用二代测序法对患儿遗传性肾病相关基因进行检测(金域检验):BBS5基因(NM_152384.2)杂合突变,突变位点为c.409T>C。采用一代测序法验证患儿母亲的BBS5基因,患儿母亲在该位点未见突变,其父未行相关检查(图3)。根据患儿男性性腺发育异常、肾脏异常2条主要体征及多饮/多尿1条次要体征,结合斜视、视物模糊、短指等表现,临床诊断为非典型Bardet-Biedl 综合征(Bardet-Biedl syndrome, BBS)。
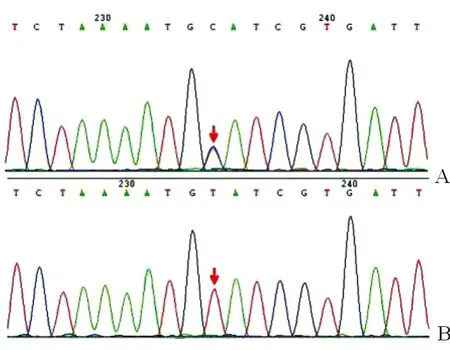
图3 患儿及其母亲BBS5基因测序
2 讨 论
BBS是一种罕见的常染色体隐性遗传病[1],于1920年首次被报道[2]。该病患病率为1/160 000~1/140 000[3],存在显著的地区差异,如科威特贝都因人患病率为1/13 500[4]。该病具有复杂的遗传背景,可累及多个器官,临床表现多样,包括视网膜营养不良、多指(趾)、肥胖、学习障碍、男性性腺发育异常、肾脏异常及言语障碍/迟缓、多尿/多饮(肾性)、共济失调/协调及平衡能力差、轻度痉挛(尤其是双下肢)、糖尿病、牙齿拥挤/小牙根/高腭弓、左心室增大/先天性心脏病、肝脏纤维化等[3-6]。各临床表现大多在出生后10年、20年相继发生。该病由于缺乏特异性的症状及体征,且发病隐匿,极易被误诊、漏诊。
BBS是常染色体隐性遗传性疾病,发病至少与21个基因突变有关,包括BB1~20及NPHP1。各基因突变在BBS患者发病过程中的作用不尽相同,具有基因异质性,但所有的BBS基因均参与纤毛功能的形成。当BBS基因突变引起所编码的蛋白质改变时,可导致纤毛的组装过程或信号转导复合物的形成受阻,从而引起相应的疾病,提示BBS是一种纤毛功能缺陷疾病[5]。本研究患儿基因检测提示,BBS5基因(NM_152384.2)杂合突变,突变位点为c.409T>C,该基因位于2q31.1。采用UCSC genome 进行比对时发现,该杂合性点突变BBS5基因409位核苷酸由T变为C,可能会导致编码氨基酸由酪氨酸变为组氨酸。查阅Ensemble数据库及ClinVar,未发现该突变位点作为单核苷酸多态性(SNP)或致病性突变位点的报道。SIFT预测该突变评分为1.0,提示很可能有害。因此,本研究患儿的致病基因突变可改变蛋白质结构,考虑为新发致病突变。
早期的观点一直未将肾脏疾病作为BBS的观察指标。但近20余年国外的长期随访结果提示,长期存活BBS患者的肾脏异常可达95%[6]。因此,在近10余年提出的BBS新的诊断标准中,将肾脏异常作为确诊的第6大主要特征,并认为肾结构和(或)功能异常是BBS常见的首发临床症状及主要死亡原因[3]。BBS诊断标准包括主要特征和次要特征,主要特征包括:视网膜营养不良、多指(趾)、肥胖、学习障碍、男性性腺发育异常和肾脏异常;次要特征包括:发育障碍/迟缓、斜视/白内障/散光、短指/并指、生长发育延迟、多尿/多饮(肾性多尿多饮)、共济失调/协调及平衡能力差、轻度痉挛(尤其指双下肢)、糖尿病、牙齿拥挤/缺牙/小牙根/高腭弓、左心室增大/先天性心脏病及肝脏纤维化。满足4个主要特征或满足3个主要特征+2个次要特征即可诊断为BBS[7]。肾脏异常包括肾结构和(或)功能异常。肾脏形态结构改变最常累及肾髓质,常见囊性变和肾发育异常,也可见肾积水、马蹄肾、异位肾、输尿管畸形、肾盏杵状畸形、胎儿性分叶肾、小肾畸形、肾憩室等。肾脏功能异常多表现为肾小管间质损伤,临床表现为多饮、多尿及尿比重下降等[8],常无血尿、蛋白尿、少尿、水肿等改变,故易被忽视、遗漏,造成早期诊断困难[9]。
根据典型的临床表现,BBS常被分为5种类型。完全型:肥胖、智力低下、视力减退、多指(趾)畸形及性腺发育;不完全型:无多指(趾)畸形或性腺发育不良;顿挫型:只有1、2项表现或几项不明显的变化;不典型型:无视网膜色素变性,可有眼部其他症状;广泛型:除完全型的5项表现外,还有其他先天异常或遗传性疾病[10-11]。在部分研究中,除了观察到上述典型的临床表现外,尚有多种不典型表现。Halac等[12]通过对Sainte-Justine医院诊断的15例BBS患者的研究发现,15例患者中有3例伴有自身免疫性疾病,其中1例伴有特发性风湿性关节炎,1例伴有1型糖尿病、桥本甲状腺炎和银屑病,1例伴有克罗恩病、原发性硬化型胆管炎及桥本甲状腺炎。这提示BBS患者可伴发慢性炎症性疾病及自身免疫性疾病,临床在对BBS患者的随访过程中应予以注意。Tica等[13]在1例22岁女性BBS患者中甚至发现卵巢畸胎瘤。
综上所述,BBS较为罕见,临床表现多样,肾脏受累近年来逐渐受到重视。该例患者无特殊家族史,家族无近亲结婚史,初次起病主要表现为多尿、多饮,但未予重视,后病情进行性加重,逐渐出现肾衰竭,辗转多家医院未查出原发病,大多数医院仅关注患儿肾衰竭的诊治,未进行原发病的筛查。随着病情进展,患儿逐渐出现多系统受累表现,包括斜视、性腺发育异常等,提示遗传性疾病可能性大。遂完善遗传性肾病相关基因检测,提示BBS5基因突变,诊断为BBS。因此,对于肾衰竭患者,寻找肾衰竭的原发病,若有多系统受累,提示遗传性疾病BBS可能,条件允许者应尽早完善相关基因检测,早期诊断、早期干预,以改善预后。
