粉煤灰磁性吸附剂的制备及磷吸附机理
2018-08-01李建军但宏兵IslamNazrul杨露敏叶先康朱金波
李建军 但宏兵 谢 蔚 Islam Nazrul 杨露敏 叶先康 朱金波
(1安徽理工大学材料科学与工程学院,淮南 232001)
(2伍伦贡大学超导与电子材料研究所,澳大利亚,伍伦贡 2500)
0 引 言
氮、磷等元素的含量超标是造成水体富营养化的主要原因[1]。研究表明,藻类等水生植物对氮、磷的需求比例符合经验公式C106H263O110Nl6P[2],即氮、磷物质的量之比为16∶1。依据Leibig最小化法则(Leibig law of the minimum)可知,磷含量对水体富营养化的影响更为敏感[3],在治理水体富营养化方面,除磷比脱氮见效更快。现有的污水除磷方法包括化学沉淀、生物转换及电解法等[4-5],这些方法虽具有一定的除磷效果,但仍存在除磷效果不稳定、污泥产生量大、适用性不广等不足。在此背景下,吸附法成为颇具潜力的除磷方法。现有磷吸附剂包括有机高分子类和无机金属氧化物及其盐类。近年来,新型无机磷吸附剂以比表面积大、吸附速度快、性能与结构稳定等特点成为研究热点[6-7],其中,含镧化合物的磷吸附性能尤为突出。镧系磷吸附剂对水中的含磷离子具有良好吸附能力,不但选择性好,而且适用于低浓度含磷离子的吸附;其吸附磷后的产物对酸碱环境适应性好,不易产生二次污染;镧离子的生物无毒性突出,有利于水资源的循环利用。镧系磷吸附剂的研究主要集中在纳米镧化合物[8]、镧化合物/非金属(高分子)复合材料[9-10]、负载型镧吸附剂等方面,其中,镧负载膨润土[11-12]研究最深入,部分产品已实现工业应用。镧负载沸石/多孔材料[12-14]及镧负载新型碳材料的研究方兴未艾,促使磷吸附性能不断提高[15-16]。为提高氧化镧/氢氧化镧的磷吸附性能,一些小组开始研究金属离子共掺杂的(La,M)Ox的镧系吸附剂[17]。
由于吸附法除磷要求吸附剂具有较大的比表面积并能较好的悬浮在水体中,因此磷吸附剂一般采用微细颗粒,由此造成吸附剂固液分离困难,此难题成为制约吸附法广泛应用的瓶颈之一。近年来,一些研究者将磁分离技术引入污水处理中,以期利用磁场力促进吸附剂的固液分离[18]。这些研究多以化学合成的纳米Fe3O4作为磁核[19-21]。由于纳米Fe3O4合成工艺较复杂、生产成本和保存条件要求高且易造成二次污染,在一定程度上限制了磁性磷吸附剂的广泛应用,因此寻找清洁廉价的磁核材料非常必要。粉煤灰磁珠来源于工业固废粉煤灰,来源广泛、价格低廉,由于其具有孔隙丰富、磁性强等特点,因而在重介质选煤、污水处理以及磁性复合材料制备等领域具有应用潜力[22-23]。如果将粉煤灰磁珠用作磁核处理则可望克服纳米磁核的不足,为磁性磷吸附剂的工业化应用铺平道路。同时,也可实现粉煤灰磁珠高附加值资源化利用,以废治废,环保效益和经济效益显著。粉煤灰磁珠作为磁核材料的主要障碍在于磁珠密度较大、颗粒较粗,因此在污水中悬浮性差、与水体作用面积不足。为此,需要对其进行预加工处理,以减小粒径、增大比表面积。
本论文以精选、球磨处理后的粉煤灰磁珠作为磁核,通过化学沉淀法,制备了氧化镧负载粉煤灰磁珠(CMS@La2O3)磷吸附剂。对所得样品的形貌、结构以及磁性进行了系统表征,并将其用于吸附磷模拟污水中的含磷离子。在研究磷吸附影响因素及吸附动力学的基础上,探讨了磷吸附反应机理。
1 实验部分
1.1 实验药品及仪器设备
实验所用粉煤灰来源于大唐淮南洛河发电厂。化学试剂磷酸二氢钾(KH2PO4),氯化镧(LaCl3),四水合钼酸铵((NH4)6Mo7O24·4H2O),抗坏血酸(C6H8O6),半水酒石酸锑钾 (KSbC4H4O7·1/2H2O),氢氧化钠(NaOH)均为分析纯,购于上海国药试剂有限公司。实验用水为自制去离子水。
使用南京博蕴通有限公司的XGB04型行星式球磨机对粉煤灰磁珠进行球磨;采用煤炭科学研究总院生产的CXG-08SD型磁选管 (磁场强度0~350 mT连续可调)分选粉煤灰磁珠。采用日本岛津XRD-6000型X射线衍射仪(XRD)分析样品的晶体结构,扫描电压40 kV,扫描电流30 mA,辐射源为Cu 靶 Kα(λ=0.154 nm),扫描角度范围 10°~80°,扫描速度2°·min-1,并通过Jade6.0软件与标准PDF卡片进行对比,确定样品特征衍射峰。采用日本JEOL公司的JSM-7001F场发射扫描电子显微镜(SEM)和能谱仪(EDS),观察样品的形貌并进行能谱分析,工作电压20 kV。利用美国康塔仪器有限公司的BET表面分析仪,通过氮气吸附检测样品的比表面积和孔径分布,脱气温度200℃。使用南京大学仪器厂生产的HH-20振动样品磁强计(VSM)测量样品的磁性。利用日本岛津UV-2600型紫外可见分光光度计检测含磷溶液的吸光度。
1.2 实验方法
1.2.1 磁珠及磁性磷吸附剂的制备
磁珠预处理包括粉煤灰精细磁选和和球磨处理两部分[24-25]。首先利用套筛对粉煤灰进行筛分,然后通过磁选管将大于100目的粉煤灰分别在300、100 mT磁场强度下进行多次磁选,获得强磁性粉煤灰磁珠。利用行星式球磨机将所得粉煤灰磁珠在转速为250 r·min-1的条件下球磨10 h,将所得磁珠微颗粒清洗并烘干备用。
通过化学沉淀法合成磁性磷吸附剂。取2.0 g磁珠微颗粒按固液比1∶100添加至200 mL 0.05 mol·L-1LaCl3溶液中,混合均匀,以 3 mL·min-1滴加1 mol·L-1NaOH溶液,调节溶液pH值至11,利用六联电动搅拌器以500 r·min-1搅拌20 h。然后利用真空抽滤或磁分离分离混合液中的固体物,利用去离子水将固体物多次清洗至中性后,110℃真空干燥。最后将粉末样品置入箱式电阻炉500℃下焙烧2 h,自然冷却后经研磨、过筛备用。
1.2.2 磷吸附试验
1.2.2.1 磷标准曲线的绘制
利用钼酸铵分光光度法检测磷含量。向7支50 mL具塞刻度管中分别加入质量浓度为2 mg·L-1的 KH2PO4溶液 0、0.5、1、3、5、10 和 15 mL, 使用去离子水稀释至标线后,分别向其中加入10%(w/w)的抗坏血酸溶液1 mL,30 s后分别加入钼酸盐溶液2 mL,混合均匀后在室温下静置15 min。利用紫外可见分光光度计在波长为710 nm处检测溶液吸光度并绘制标准曲线。
1.2.2.2 磷吸附试验
称取0.1 g磁性磷吸附剂添加至100 mL 20 mg·L-1KH2PO4溶液,调节溶液 pH=3,置于冷冻摇床25℃条件下,以160 r·min-1恒温搅拌3 h,最后检测上清液吸光度,并与标准曲线对比,计算磷的比吸附量。比吸附量的计算公式为:

式中,qt为比吸附量(mg·g-1),C0和 Ce分别为吸附前和吸附平衡时溶液中磷的浓度(mg·L-1),V为溶液体积(L),m为吸附剂的投加量(g)。
1.2.2.3 吸附剂再生
将吸附饱和的磁性磷吸附剂置于1 mol·L-1NaOH 溶液中,常温下以 500 r·min-1解吸 3~6 h,磁分离其中的固体颗粒,洗涤至中性并充分干燥备用。再生吸附剂的磷吸附试验步骤同(1.2.2.2)所述。
2 结果与讨论
2.1 磁性磷吸附剂的结构与性能表征
2.1.1 磁性磷吸附剂的结构表征

图1 (a)CMS、(b)磁珠微颗粒、(c)CMS@La2O3的SEM图;图(d)为图(c)对应的EDS能谱;图(e~h)为图(c)对应的EDS面扫描图像Fig.1 SEM images of(a)CMS,(b)CMS microparticles and(c)CMS@La2O3;(d)Corresponding EDS spectrum of CMS@La2O3in(c);(e~h)Corresponding EDS surface scanning images of CMS@La2O3in(c)
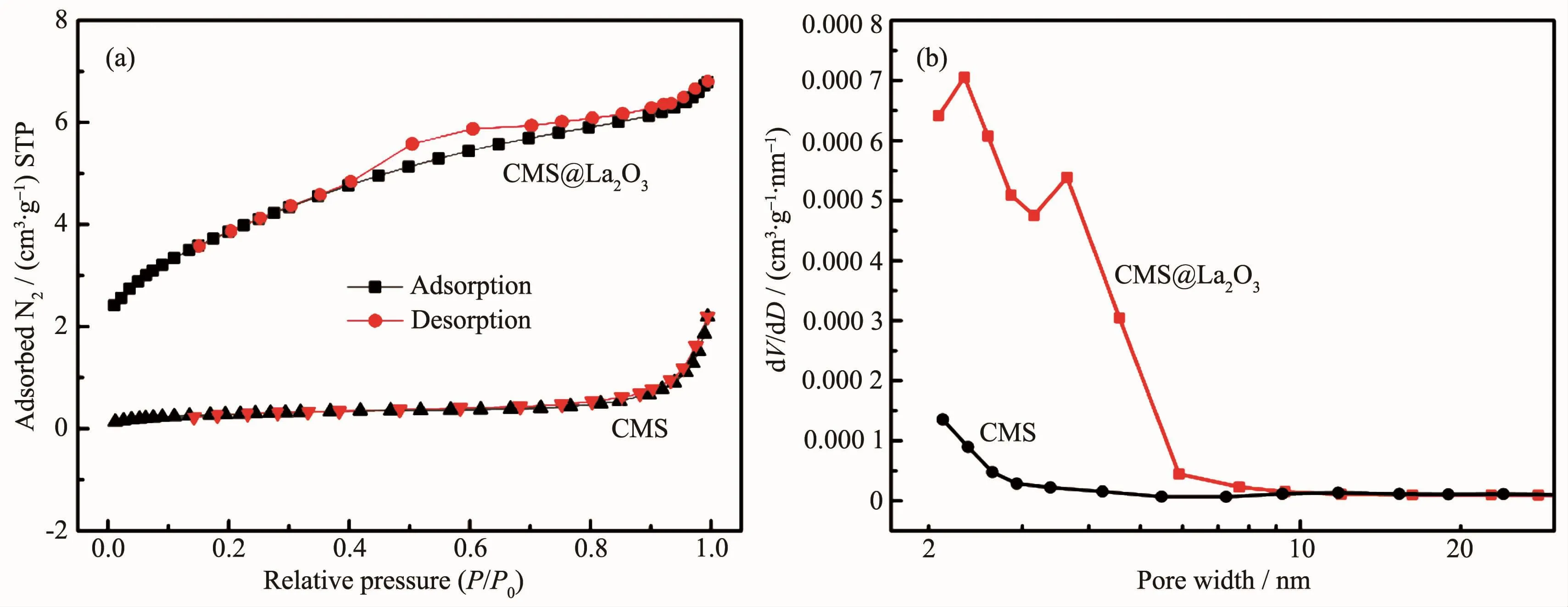
图2 CMS和CMS@La2O3的(a)N2吸附-脱附曲线和(b)孔径分布曲线Fig.2 (a)N2adsorption-desorption isotherm and(b)pore size distribution of CMS and CMS@La2O3
由样品的SEM图(图1)可知,经球磨破碎后,磁珠颗粒的平均粒径减小约1个数量级,形状由类球形变成了不规则形状。而经镧改性后,磁珠颗粒表面有白色结晶物质生成,因此变得较为粗糙。EDS分析表明,改性后磁珠的能谱中出现了较强的镧元素峰,镧元素质量百分数可达28.76%。面扫描能谱图表明,镧改性样品中除磁珠原有的O、Fe、Si、Al等元素外,广泛分布有镧元素,且在SEM图中白色结晶物质对应位置上的镧元素含量最高。说明磁珠表面的凸起包覆物应为镧化合物,镧元素较均匀地包覆在磁珠微颗粒表面。BET氮气吸附结果显示(图2a),经氧化镧包覆后,样品的比孔隙率大幅增加,从磁珠微颗粒原有的2 cm·g-1增加到镧包覆样品的7 cm·g-1,孔径分布图(图2b)表明氧化镧包覆层具有丰富的孔隙结构,主要为孔径低于6 nm的微介孔,这些孔隙结构对于提高样品的物理吸附能力是有利的。
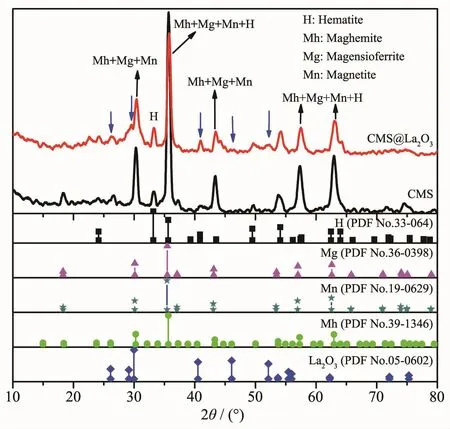
图3 CMS和CMS@La2O3的XRD图Fig.3 XRD patterns of CMS and CMS@La2O3
为确定样品中镧化合物的结构,对样品进行了XRD分析。由图3可知,原粉煤灰磁珠中含有磁铁矿(magnetite)、镁铁矿(magnesioferrite)、赤铁矿(hematite)和磁赤铁矿(maghemite)等结构。CMS@La2O3的XRD图中保留了原磁珠的所有衍射峰,说明改性过程未改变磁珠的结构。但多数衍射峰峰型有所宽化、强度有所下降,这是由于镧化合物包裹在磁珠表面所致。值得注意的是,CMS@La2O3的XRD图中在 27.18°、29.96°、40.54°、45.31°和 53.77°出现了 5个新的衍射峰,如图中蓝色箭头所示,分别对应于氧化镧(PDF No.05-0602)的(100)、(101)、(102)、(110)和(103)晶面。这说明磁珠微颗粒表面生成了氧化镧晶体,与SEM及EDS表征结果相符。2.1.2 磁性吸附剂的磁性
图4(a)是镧改性前后磁珠的磁滞回线。由图可知,磁珠微颗粒的比饱和磁化强度为39.80 emu·g-1,而CMS@La2O3的比饱和磁化强度为20.35 emu·g-1。这是由于经过氧化镧负载后,样品中磁性物质相对含量减少。虽然CMS@La2O3的磁性有所减弱,但仍具有较强铁磁性,如图4(b)所示,永磁体在距溶液2 cm外即可对CMS@La2O3实现高效磁分离。
2.2 磷吸附性能研究
2.2.1 磷标准曲线
由图5可知,在波长为710 nm处测定磷的标准曲线:y=1.861 5x+0.005 7,其中,R2=0.999 6,这说明在质量浓度为0~0.6 mg·L-1的测量范围内,定波长吸收强度与磷浓度具有较好的线性关系。
2.2.2 磷吸附量随时间的变化
利用所得磁珠微颗粒和CMS@La2O3磁性吸附剂对P模拟污水进行吸附处理,试验结果如图6所示。磁珠微颗粒本身对含磷离子的吸附能力很弱,其饱和比吸附量只有2.07 mg·g-1;而经La2O3包覆后磷吸附能力显著增强,比吸附量最高可达19.50 mg·g-1。从吸附曲线上看,CMS@La2O3对磷的比吸附量随吸附时间的延长而逐渐增加,在磷吸附的初期,吸附速度很快,10 min内磷的去除率即可超过50%;当吸附时间超过60 min后,磷吸附速度迅速减缓,3 h后趋于饱和。与同类磁性磷吸附剂相比,CMS@La2O3吸附剂的磷比吸附量处于中等水平,具体吸附性能及吸附动力学模型对比如表1所示[9,13,18,26-31]。值得注意的是,该吸附剂在磷吸附过程中,溶液pH值随着磷比吸附量增加的同时也在不断增大。pH值增大说明在磷吸附过程中不断产生氢氧根,意味着CMS@La2O3对含磷离子的吸附可能源于La2O3表面离子的配位不饱和引发的化学吸附[32]。La2O3表面与水分子配位-反应的过程如图7所示,表面La离子首先与水分子配位,水分子的离解性化学吸着导致表面羟基化。羟基化的表面可与溶液中的磷酸根、磷酸氢根、磷酸二氢根等含磷离子发生离子交换,并释放出氢氧根。其反应方程式如下:
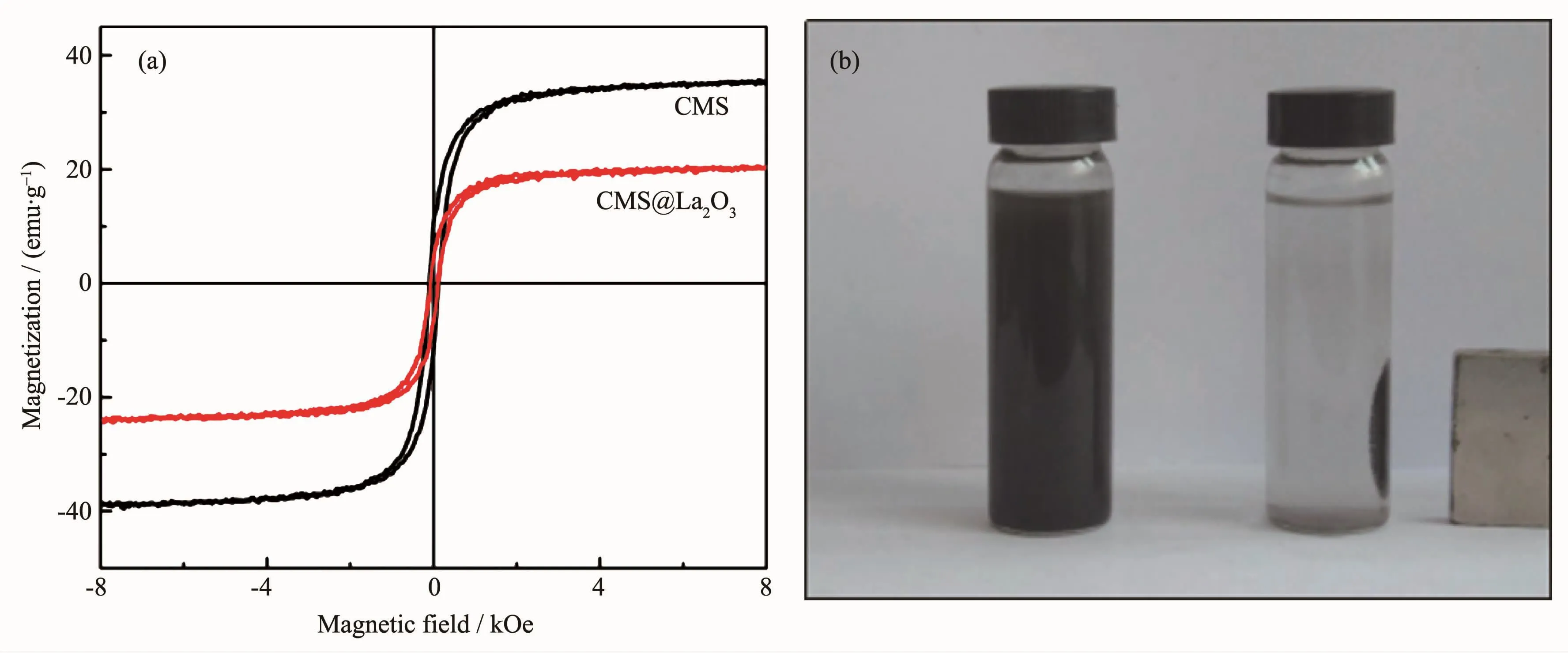
图4 (a)CMS和CMS@La2O3的磁滞回线;(b)CMS@La2O3颗粒被永磁体吸引分离Fig.4 (a)Magnetic hysteresis loops of CMS and CMS@La2O3;(b)CMS@La2O3particles are separated by the permanent magnet
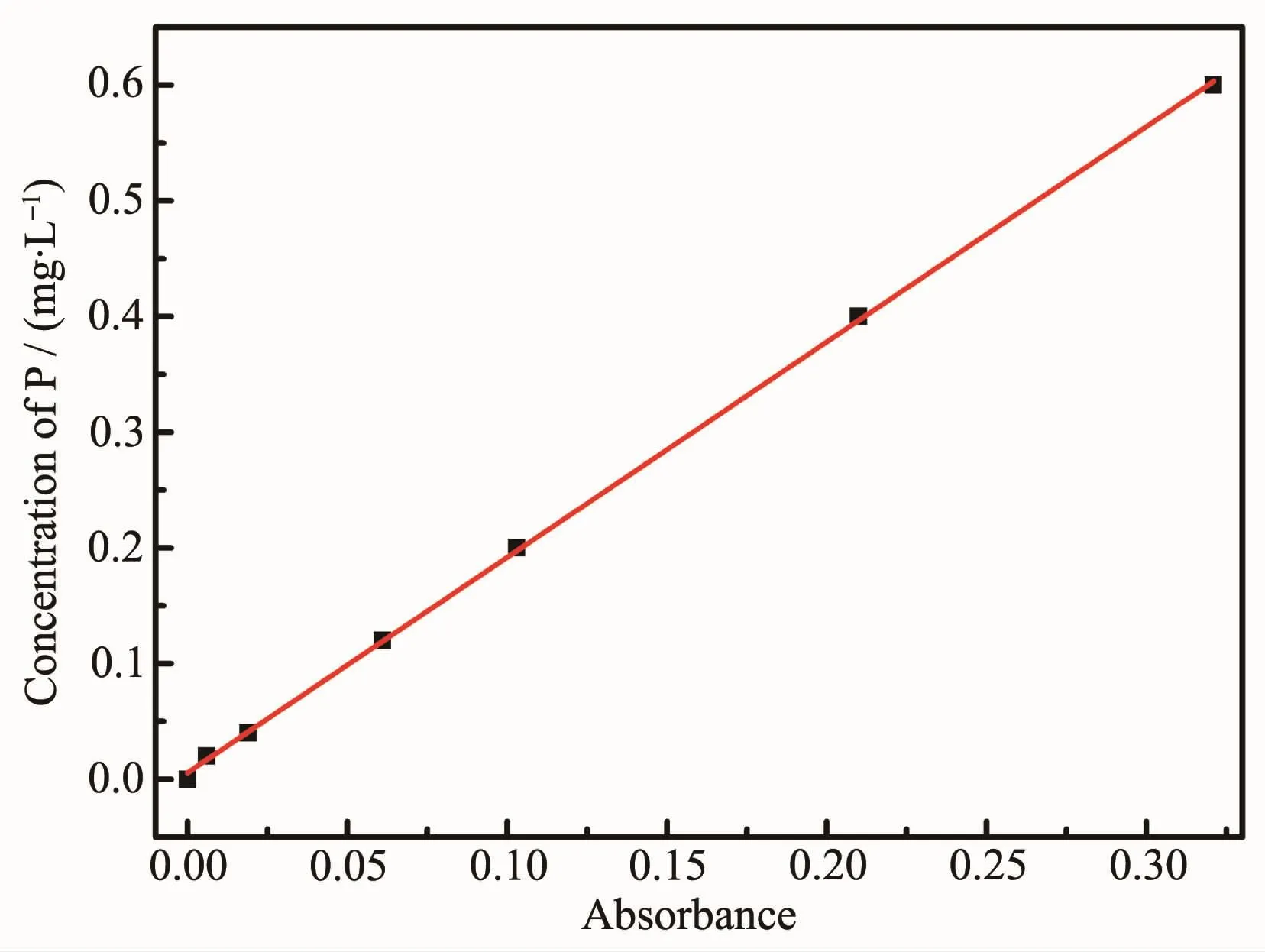
图5 磷的浓度标准曲线Fig.5 Standard curve of P concentration
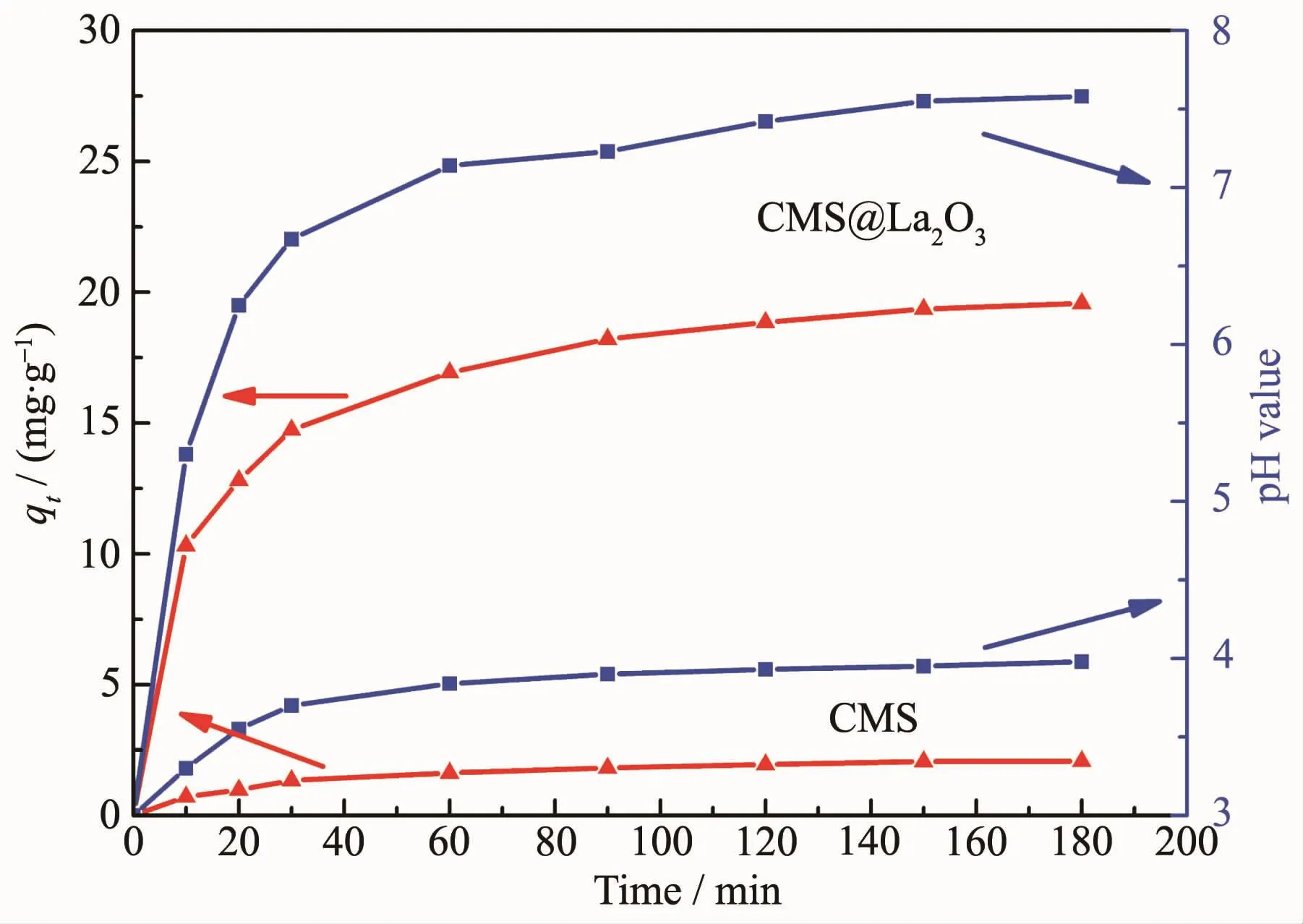
图6 吸附时间对CMS和CMS@La2O3磷吸附性能的影响Fig.6 Effect of adsorption time on the P adsorption properties of CMS and CMS@La2O3
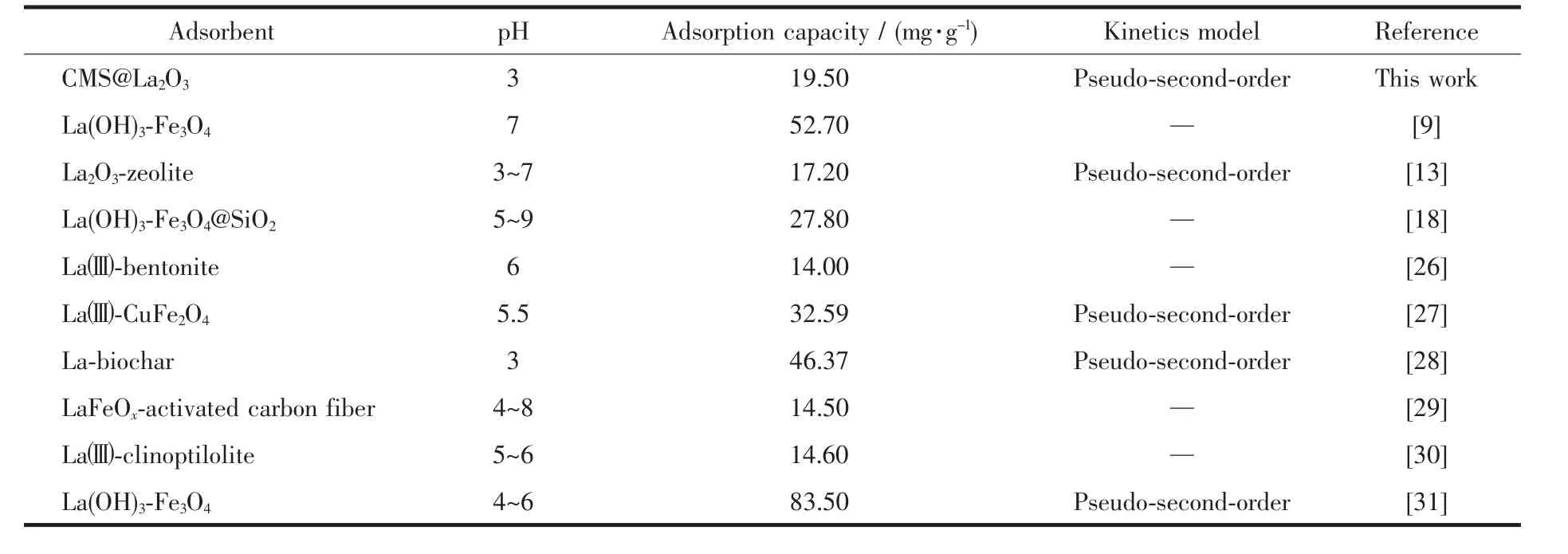
表1 部分镧系磷吸附剂的吸附性能及动力学模型对比Table 1 Comparison of adsorption performance and kinetics model of reported Lanthanide phosphorus adsorbents
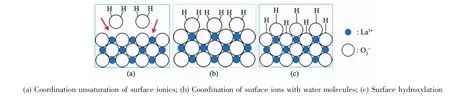
图7 La2O3表面与水分子配位-反应的机理示意图Fig.7 Schematic diagram of the coordination reaction between the surface of La2O3and water molecules
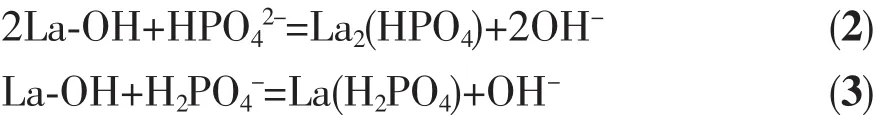
2.2.3 溶液pH值对磷吸附的影响
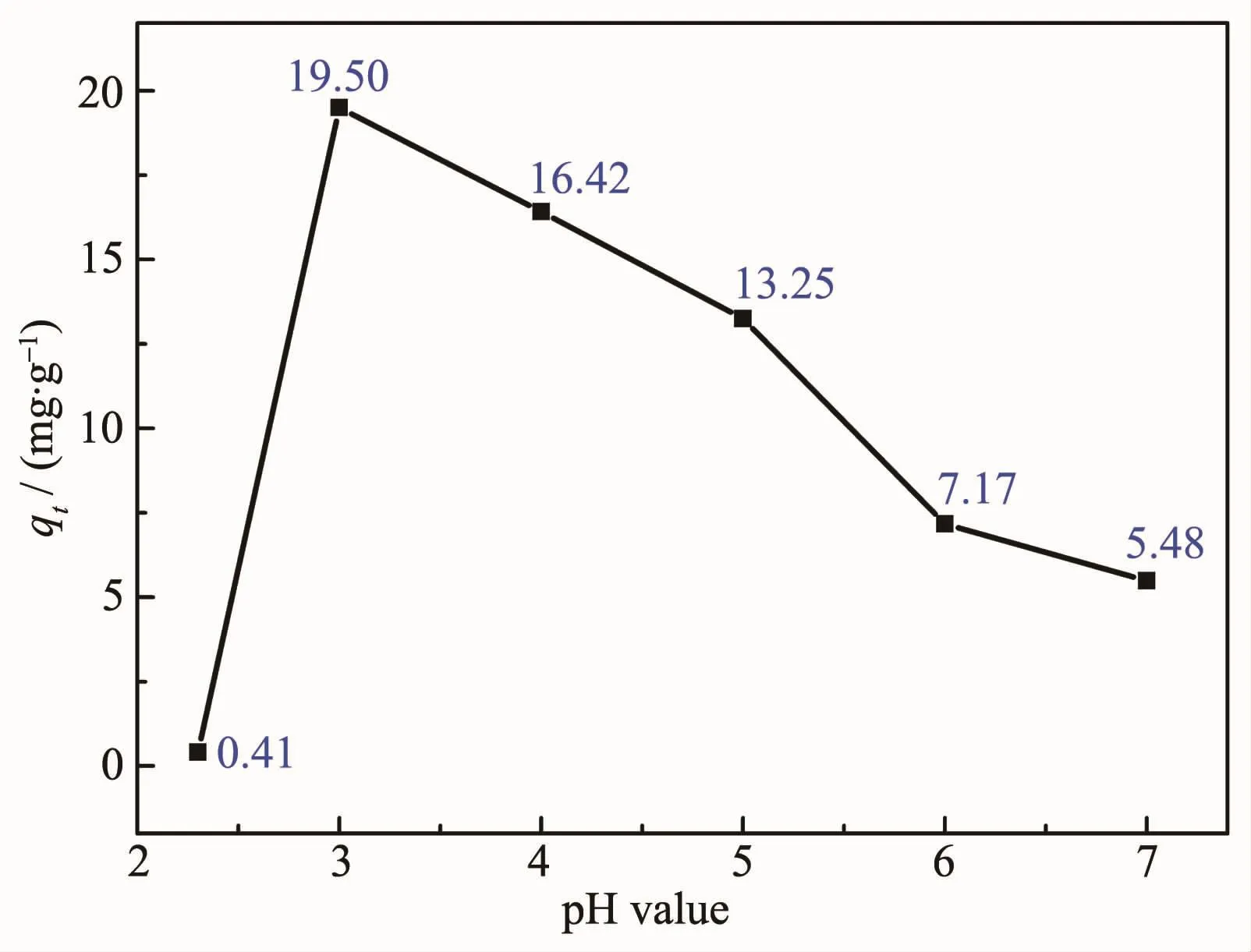
图8 溶液pH值对CMS@La2O3磷吸附率的影响Fig.8 Effect of solution pH value on the P adsorption capacity of CMS@La2O3
图8 为溶液初始pH值对磷吸附率的影响曲线。由图可知,从中性水开始,磷的比吸附量随pH值的降低而逐渐增加,当溶液pH值为3时,比吸附量达到最高;但当溶液pH值小于3时,磷吸附量迅速下降。含磷污水中的OH-会与含磷离子竞争吸附剂表面的活性位点[33],当溶液pH值不断减小时,该竞争作用就会减弱,因而磷吸附不断增强。同时,酸性增强会中和磷吸附反应中反应产物OH-,促进公式(2)、(3)反应的进行。此外,在不同pH值下磷酸盐的种类也会发生变化,酸性环境下的磷酸盐对氧化镧的亲和力较强[34-35],故在偏酸性的溶液中含磷离子的比吸附量要高于偏碱性的溶液环境。当溶液pH值低于3时,磷比吸附量迅速下降是因为此时溶液酸性过强,溶液中溶解平衡占据主导地位,镧的羟基化合物趋于溶解[36-37],不利于反应(2)、(3)的进行,难以对磷酸根产生固定化作用[36]。此外,有研究表明,强酸性条件下不利于磷酸根吸附[33]。2.2.4 共存阴离子对磷吸附的影响
为探究溶液中常见阴离子对CMS@La2O3磷吸附的影响,分别做了不同浓度的氯离子、硫酸根和碳酸根干扰对比实验。如图9所示,不同阴离子的存在对CMS@La2O3的磷吸附效果的影响显著不同。当KH2PO4污水中存在Cl-离子时,在 0~0.20 mol·L-1的浓度范围内,磷的比吸附量波动范围小于0.6 mg·g-1,说明Cl-离子对磷吸附干扰很小。而CO32-和SO42-离子对磷吸附的影响很显著,当二者掺入离子浓度达到0.05 mol·L-1时,磷比吸附量分别下降8.38 和 6.61 mg·g-1。 这是由于 CO32-和 SO42-干扰离子与含磷离子之间对吸附位点产生了竞争吸附[38]。当干扰离子占据了氧化镧表面的部分吸附位点,就会造成含磷离子吸附位点数不足、磷比吸附量下降。此外,CO32-在溶液中会水解释放大量的氢氧根[39],氢氧根也会竞争吸附CMS@La2O3表面的活性位点。同时,La2(CO3)3的溶度积常数(3.98×10-34)低于LaPO4的溶度积常数(3.7×10-23)[40],说明 La2(CO3)3比LaPO4更难溶,故在溶液中,部分LaPO4会向La2(CO3)3转化,从而释放出部分磷酸根,使磷吸附量降低,故CO32-对CMS@La2O3磷吸附的抑制作用最强,与之前的研究结论相吻合[39-41]。与Cl-相比,当SO42-不断积聚在CMS@La2O3吸附剂表面时,与磷酸根离子具有更强的静电排斥作用[42],因此SO42-比Cl-具有更强的磷吸附干扰作用。
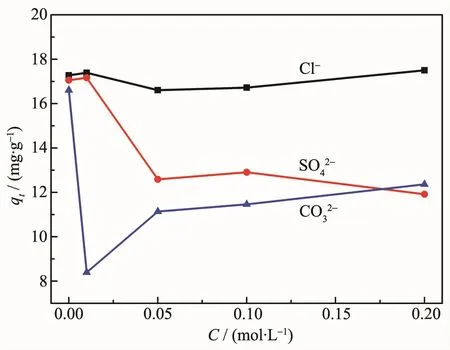
图9 共存阴离子浓度(C)对CMS@La2O3磷吸附量的影响Fig.9 Effect of concentration of coexisting anions(C)on the P adsorption capacity of CMS@La2O3
2.3 吸附动力学研究
用于研究离子描述液-固吸附过程的动力学模型包括准一级与准二级动力学模型。其数学表达式为:
准一级动力学模型:

准二级动力学模型:

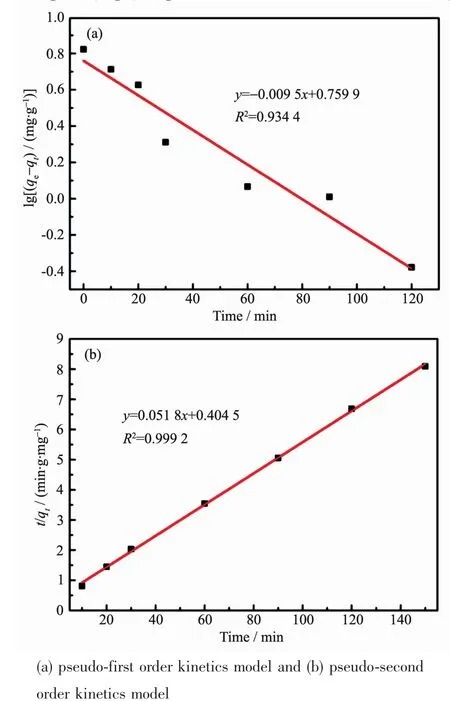
图10 CMS@La2O3的动力学模型Fig.10 Kinetics model of CMS@La2O3
式中:qe与qt分别为吸附平衡时和吸附时间t时的比吸附量(mg·g-1),t为吸附时间(min),k1与 k2为吸附速率常数(min-1,g·mg-1·min-1)。
利用2种动力学模型对CMS@La2O3除磷过程进行拟合,如图10所示。由图可知,准一级方程的拟合相关系数R2为0.934 4,准二级方程的拟合相关系数R2为0.999 2。显然准二级动力学模型的拟合相关系数R2更接近于1,而且由准二级方程计算得到的qe值(19.31 mg·g-1)也与实验值(19.50 mg·g-1)更接近,以上结果表明准二级方程能更好地描述磷在氧化镧负载粉煤灰磁珠表面的吸附行为,即CMS@La2O3吸附剂的磷吸附过程以化学吸附为主。由于离子交换作用属于化学吸附过程,间接印证了前述公式(2)、(3)的磷吸附反应机理,说明CMS@La2O3的除磷机理为La2O3表面的羟基化-离子交换。
2.4 吸附剂的再生
吸附磷后的CMS@La2O3,可通过处理实现再生循环利用。具体方法可简述为,将CMS@La2O3磁分离,置于 1 mol·L-1NaOH 溶液中,剧烈搅拌 3~6 h,然后清洗至中性,干燥后即可回用。如图11所示,再生CMS@La2O3磷吸附剂前期的吸附速度依然很快,2 h后吸附趋于饱和,初次回收利用磷吸附剂的比吸附量为18.43 mg·g-1,可达原吸附剂的94.62%。随着再生次数的增加,磁性吸附剂的磷比吸附量逐渐降低。
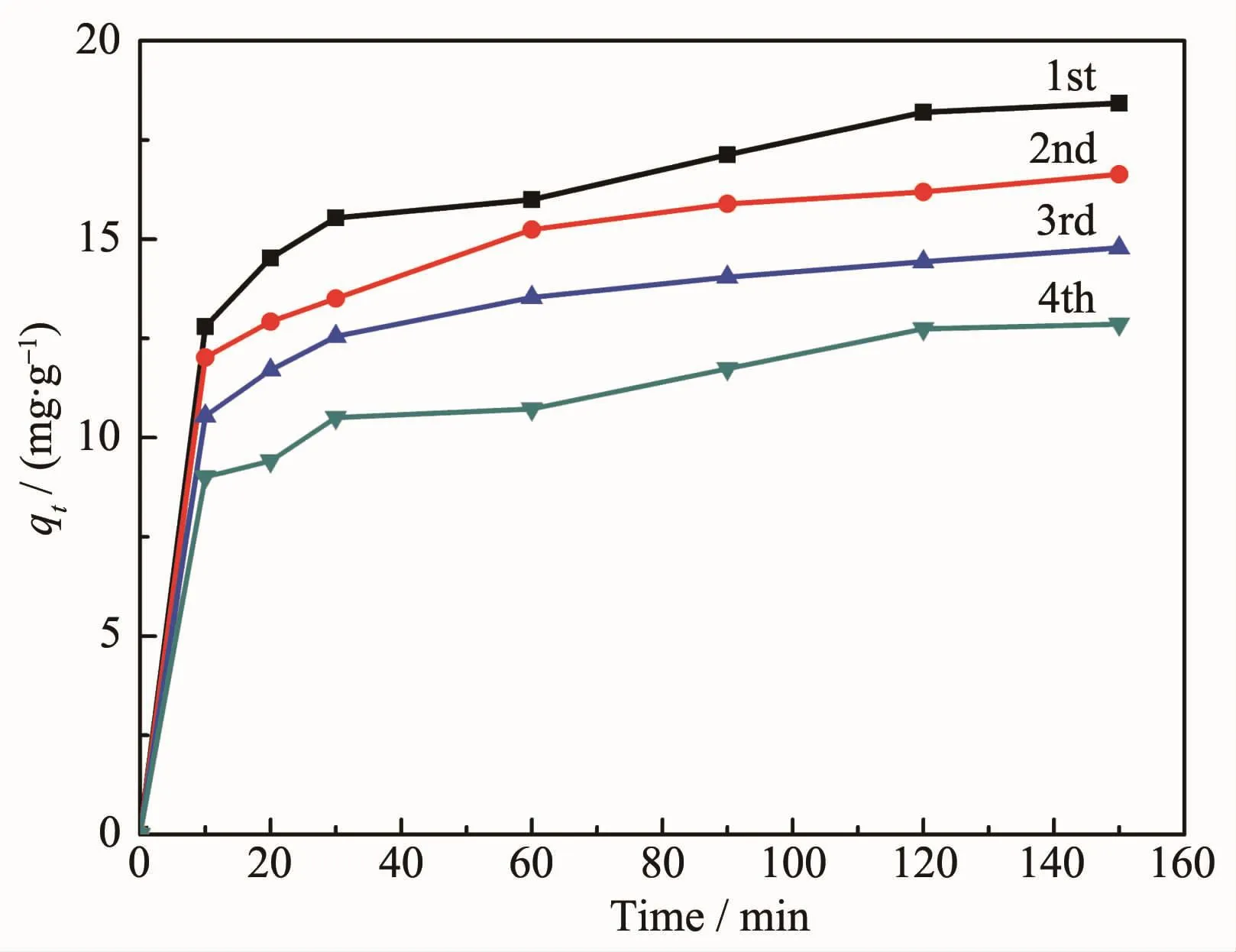
图11 再生CMS@La2O3吸附剂的磷吸附曲线Fig.11 P adsorption curves of regenerated CMS@La2O3 adsorbent
3 结 论
(1)以粉煤灰磁珠微颗粒为磁核,通过化学沉淀法制备了CMS@La2O3磁性磷吸附剂,其比磁化强度可达20.35 emu·g-1,在外加磁场作用下,可实现高效磁分离。
(2)磷吸附试验表明,当投加1 g·L-1CMS@La2O3磁性磷吸附剂处理pH值为3、浓度为20 mg·L-1的含磷污水时,其最高比吸附量为19.50 mg·g-1。CMS@La2O3吸附剂的磷吸附性能与吸附时间、溶液pH值以及共存阴离子有关。使用过的吸附剂经处理后可多次循环利用。
(3)CMS@La2O3的磷吸附过程符合吸附动力学准二级方程,以化学吸附为主。吸附反应机理为La2O3表面羟基化-离子交换。
